Synthesis, Characterization and In Silico Studies of Novel (E)-4-(((3-(substituted phenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide as Diuretic Agents
Viswaskumar Panchal1* , Zakirhusen Gadhawala1, Arun Malaviya1 and Shreekant Prajapati2
, Zakirhusen Gadhawala1, Arun Malaviya1 and Shreekant Prajapati2
1Department of Chemistry, The HNSB. Ltd. Science College, Himatnagar, Gujarat, India, (affiliated to Hemchandracharya North Gujarat University, Patan), India.
2Department of Chemistry, Hemchandracharya North Gujarat University, Patan, India.
Corresponding Author E-mail: vishwas.edu@hotmail.com
DOI : http://dx.doi.org/10.13005/ojc/400212
Article Received on : 01 Dec 2023
Article Accepted on :
Article Published : 27 Mar 2024
Reviewed by: Dr. kumaran R.
Second Review by: Dr. Rajesh Chawla
Final Approval by: Dr. Dinesh Chand Sharma
This research delves into the examination of benzene sulphonamide derivatives featuring pyrazole rings as potential diuretics. Concentrating on their role as human carbonic anhydrase inhibitors (hCA), the investigation aims to unveil a groundbreaking diuretic drug. Six innovative benzenesulfonamide derivatives are synthesized utilizing a conventional heating process. Subsequently, employing AutoDock Vina 1.2.3, these compounds undergo molecular docking assessments and pharmacokinetic predictions at the active sites of hCA I and hCA II, while the SwissADME program is employed for pharmacokinetic forecasting. Notably, Compounds 17 and 19 exhibit robust binding affinities with hCA I and II, respectively, as evidenced by the docking study. ADME studies reveal favorable bioavailability and adherence to PAINS alerts, as well as Lipinski's rule of five requirements. Consequently, based on the findings, these compounds exhibit significant potential as diuretics in comparison to well-established acetazolamide medications.
KEYWORDS:ADME; Benzenesulfonamide; Carbonic anhydrase Docking; Diuretic; Pyrazole
Download this article as:| Copy the following to cite this article: Panchal K, Gadhawala Z, Malaviya A, Prajapati S. Synthesis, Characterization and In Silico Studies of Novel (E)-4-(((3-(substituted phenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide as Diuretic Agents. Orient J Chem 2024;40(2). |
| Copy the following to cite this URL: Panchal K, Gadhawala Z, Malaviya A, Prajapati S. Synthesis, Characterization and In Silico Studies of Novel (E)-4-(((3-(substituted phenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide as Diuretic Agents. Orient J Chem 2024;40(2). Available from: https://bit.ly/3PD2Pa3 |
Introduction
A class of drugs known as diuretics increases the flow rate of urination1, which in turn causes the body to generate more urine. Diuretics increase the excretion of water and electrolytes, especially salt and chloride ions, without affecting the absorption of protein, vitamins, carbohydrates, or amino acids. Diuretics are used to treat a variety of edematous disorders, such as nephrosis and congestive heart failure2. They can also be used as an adjuvant therapy for cataracts, acute mountain sickness, hypercalcemia, and primary hyperaldosterism3. Diuretics have been used in sports where quick weight reduction is necessary to achieve a weight category because of their strong capacity to drain water from the body4.Within the area of sports medicine, diuretics were initially prohibited by the International Olympic Committee (IOC) in 1988 for the Calgary Winter Olympics, Despite this restriction, diuretics are still included among the chemicals and procedures used in doping; In sports where weight categories are involved, diuretics may be abused for two primary purposes: (i) to accomplish rapid weight reduction prior to competition, and (ii) to conceal the use of other doping drugs by changing their excretion mechanism, primarily by decreasing their concentration in urine 5–7. Diuretic medications fall into many categories: thiazides (like benzthiazide), potassium sparing diuretics (like amiloride), loop diuretics (like bumetanide), carbonic anhydrase inhibitors (like acetazolamide), mercurial diuretics (like mersalyl) and osmotic diuretics (like mannitol) 4.In situations of overdose or poisoning, certain diuretics, Moreover, medications like acetazolamide serve to alkalinize urine and quicken the removal of acidic substances. Although diuretics work on several nephron sites to create their effects, one of the main mechanisms is the reduction of sodium ion reabsorption throughout the renal tubules of the kidney. 8–10. Zinc-containing enzymes known as “metalloenzymes,” or carbonic anhydrases (CAs), catalyze the interconversion of CO2 and H2O into bicarbonates (HCO3-), using a metal hydroxide and nucleophilic mechanism 1,11–13. There are eight different kinds of carbonic anhydrases that are known, but only the α-type, which has 16 isozymes, is prevalent in humans 14. The α-type CAs differ in how these enzymes prefer the zinc ions (within the active site) that catalyzes reactions 11. Studies on the catalytic activity of CAs and their inhibition or activation are essential for the treatment of many disorders that are clinically significant 15,16.
The pyrazole scaffold is a flexible molecule that has garnered interest in this context because of its broad range of varied pharmacological properties. As a result, it is a versatile lead molecule in numerous authorized medications, including rimonabant, lonazolac, ramifenazone, and celecoxib 17–20. Pyrazoles have a number of significant biomedical characteristics 21. Numerous medicinal properties, including insecticidal22, acaricidal23, anticonvulsant24, antidepressant25, antiulcer, diuretics and anticancer properties26, are displayed by this class of compounds. Despite the fact that the inhibition of hCAs by aromatic and heterocyclic sulfonamides has been extensively researched over the past 50 years and used to treat a variety of illnesses 27,28. In the active region of the CA enzyme, the main sulfonamide group binds to the Zn2+ ion as an anion (SO2NH−), preventing catalysis 1,29. The absence of selectivity, which may cause harmful side effects, hinders the creation of efficient CA inhibitors. Thus, we have been interested in creating hCA inhibitors that are not only strong but also show good selectivity for a particular isoform. The synthesis of some pyrazole derivatives with sulfonamide moiety and their capacity to inhibit hCA I and II isoforms was the main focus of our research in the current work. Computational studies were also used to shed light on their effects based on the binding interactions with the target enzymes and their predicted molecular properties.
Material and Methods
Without undergoing additional purification, all the compounds were acquired from the commercial providers Merck and thermo fisher scientific. Dimethylformamide (DMF), n-hexane, ethanol, methanol, and ethyl acetate were among the solvents used. Phenylhydrazine, 4-Flouroacetophenone, 3,4-Chloroacetophenone, 3-Bromoacetophenone, 4-Choloracetophenoe, 4-Bromoacetophenone, 2-Chloroacetophenone, 4-aminobenzenesulfonamide, and phosphorus oxychloride were all used in the synthesis. Using a FT-IR Spectrometer, FT-IR spectra encompassing the 400–4000 cm-1 range were acquired. NMR spectra were obtained using an Agilent 400 MR instrument running at 400 MHz for 1H NMR and 13C NMR. An LC-MS from waters was used to record the mass spectra. To track the course of reactions and evaluate the purity of the products, TLC plates (Merck Silica Gel 60 F254) were used.
Synthesis of (1E)-1-[1-(Substituted Phenyl)ethylidene]-2-phenylhydrazone(3-8)
20 ml of methanol was used to dissolve a mixture that contained 10 mmol of different substituted acetophenones, phenylhydrazone, and 1 ml of glacial acetic acid as a catalyst. At 500C, the solution was stirred for three hours. The reaction mixture was placed onto ice once TLC had confirmed that the reaction had finished. After filtering and a water wash, the resultant hydrazone intermediates 3-8 were refined by recrystallization in ethanol 30–32.
Synthesis of 3-(Substituted phenyl)-1-phenyl-1H-pyrazole-5-carbaldehyde (9-14)
The Vilsmeier-Haack reagent was made by progressively adding 1.8 ml of POCl3 to 10 ml of DMF that had been refrigerated on ice. A Hydrazone (3–8, 10 mmol) was then added. At 60°C, the resultant mixture was stirred for four hours. Following TLC verification of the reaction’s completion, the mixture was placed on crushed ice and neutralized with a bicarbonate solution. Crystallization from ethanol was used to separate and further purify the crude product 30–34.
Synthesis of E)-4-(((3-(substituted phenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide(15-20)
In order to create benzene sulfonamide Schiff bases with a pyrazole moiety, ethanol (20 ml) was used to dissolve aldehyde derivatives containing pyrazole (9-14, 10 mmol). Following that, dropwise additions of these solutions were made to solutions containing the equivalent 10-mmol 4-aminobenzenesulfonamide dissolved in 20 ml of methanol. After adding a catalytic quantity of acetic acid and stirring the reaction mixture for 24 hours at 80°C, TLC was used to track the reaction progress. The solvent was removed once the reactions were judged to be finished, and the solid was then cleaned with ice-cold ethanol After that, the resulting compounds were recrystallized from methanol/ethanol 31,35.
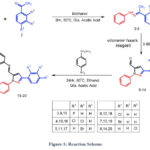 |
Figure 1: Reaction Scheme. |
(E)-4-(((3-(4-fluorophenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide(15)
White powder; 75%, MP: 198-200ºC; IR (KBr, cm–1) ν: 1619 (C=N), 1315 (Asymmetric, S=O), 1144 (symmetric, S=O); 1H NMR (400 MHz, DMSO-d6) δ: 7.29-7.37 (5H, m), 7.41-7.44 (2H, t) 7.55-7.59 (2H, t), 7.82-7.84 (2H, d), 7.92-7.94 (2H, q), 8.08-8.09 (2H, d), 8.56 (1H, s), 9.23 (1H, s); 13C NMR(100 MHz, CDCl3) δ: 118.56, 119.17, 119.99, 122.05, 122.81, 124.29, 128.12, 129.95, 130.35, 130.98, 131.46, 131.68, 134.25, 139.02, 145.53, 152.39, 154.47; ESI–MS (m/z): Calcd.: 420.46, found: 421.20 [M+H]+ for C22H17FN4O2S
(E)-4-(((3-(3,4-dichlorophenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide(16)
White powder; 80%, MP: 218-220ºC; IR (KBr, cm–1) ν: 1627 (C=N), 1320 (Asymmetric, S=O), 1149 (symmetric, S=O); 1H NMR (400 MHz, DMSO-d6) δ: 7.298 (2H, s), 7.37-7.41 (1H, t), 7.45-7.476 (2H, d), 7.52-7.56 (2H, t), 7.70 (2H, s), 7.74-7.76 (2H, d), 7.95-7.96 (2H, d), 8.20 (1H, s), 8.29 (1H, s), 8.95 (1H,s); 13C NMR(100 MHz, CDCl3) δ:119.23, 120.01, 121.77, 126.11, 127.32, 129.74, 129.91, 130.24, 130.42, 131.31, 131.73, 139.52, 142.03, 144.41, 151.98, 155.18; ESI–MS (m/z): Calcd.: 471.36, found: 472.40 [M+H]+ for C22H16Cl2N4O2S
(E)-4-(((3-(3-bromophenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide(17)
White powder; 77%, MP: 213-215ºC; IR (KBr, cm–1) ν: 1621 (C=N), 1314 (Asymmetric, S=O), 1144 (symmetric, S=O); 1H NMR (400 MHz, DMSO-d6) δ: 7.34 (2H, s), 7.35-7.38 (2H, d), 7.40-7.44 (1H, t), 7.46-7.50 (1H, t), 7.55-7.59 (2H, t), 7.66-7.68 (1H, d), 7.83-7.85 (2H, d), 7.89-7.91 (1H, d), 8.03-8.05(2H, d), 8.15 (1H, s), 8.56 (1H, s), 9.24 (1H, s) ; 13C NMR(100 MHz, CDCl3) δ: 118.75, 119.53, 122.09, 125.92, 126.91, 127.64, 129.61, 129.81, 129.99, 130.71, 130.85, 131.44, 134.65, 139.13, 142.20, 145.01,150.10, 153.37; ESI–MS (m/z): Calcd.: 480.03, found: 481.20 [M+H]+ for C22H17BrN4O2S
(E)-4-(((3-(4-chlorophenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide(18)
White powder; 69%, MP: 205-207ºC; IR (KBr, cm–1) ν: 1623 (C=N), 1315 (Asymmetric, S=O), 1147 (symmetric, S=O); 1H NMR (400 MHz, DMSO-d6) δ: 7.31 (2H, s), 7.39-7.43 (1H, t), 7.45-7.47 (2H, d), 7.53-7.56 (4H, t), 7.64-7.66 (2H, d), 7.75-7.77 (2H, d), 7.96-9.78 (2H, d), 8.21 (1H, s), 8.97 (1H, s); 13C NMR(100 MHz, CDCl3) δ: 119.69, 122.03, 125.52, 126.93, 129.95, 130.11, 130.98, 131.46, 131.72, 138.24, 142.10, 144.54, 150.88, 153.69; ESI–MS (m/z): Calcd.: 436.08, found: 437.60 [M+H]+ for C22H17ClN4O2S
(E)-4-(((3-(4-bromophenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide(19)
White powder; 72%, MP: 210-212ºC; IR (KBr, cm–1) ν: 1621 (C=N), 1315 (Asymmetric, S=O), 1146 (symmetric, S=O); 1H NMR (400 MHz, DMSO-d6) δ: 7.34 (2H, s), 7.36-7.38 (2H, d) 7.39-7.43 (1H, t), 7.51-7.62 (4H, m), 7.70-7.72 (2H, d), 7.82-7.85 (2H, m), 8.02-8.04 (2H, d), 8.55 (1H, s), 9.24 (1H, s) ; 13C NMR(100 MHz, CDCl3) δ: 119.69, 122.03, 125.52, 126.93, 129.95, 130.11, 130.98, 131.46, 131.72, 138.24, 142.10, 144.54, 150.88, 153.69; ESI–MS (m/z): Calcd.: 480.03, found: 481.20 [M+H]+ for C22H17BrN4O2S
(E)-4-(((3-(2-chlorophenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide(20)
White powder; 60%, MP: 186-188ºC; IR (KBr, cm–1) ν: 1627 (C=N), 1318 (Asymmetric, S=O), 1149 (symmetric, S=O); 1H NMR (400 MHz, DMSO-d6) δ: 7.42-7.51 (4H, m), 7.56-7.66 (5H, m) 7.69-7.71 (2H, d), 7.96-7.98 (2H, d), 8.05-8.07 (2H, d), 8.56 (1H, s), 9.23 (1H, s); 13C NMR(100 MHz, CDCl3) δ: 118.56, 119.17, 119.99, 122.05, 122.81, 124.29, 128.12, 129.95, 130.35, 130.98, 131.46, 131.68, 134.25, 139.02, 145.53, 152.39, 154.47; ESI–MS (m/z): Calcd.: 436.08, found: 437.60 [M+H]+ for C22H17ClN4O2S
Molecular Docking
The Autodock vina software 1.2.336,37 version was used for all phases of the molecular docking research, including protein and ligand preparation, active site grid creation, and ligand docking. The protein data bank (PDB) provided the 3D structures of the target proteins Crystal Structure Of Sulfonamide Drug Complexed With Human Carbonic Anhydrase I (PDB ID: 1AZM, Resolution: 2.00 Å) and Crystal Structure Of acetazolamide Drug Complexed With Human Carbonic Anhydrase II (PDB ID: 3HS4, Resolution: 1.10 Å). Using the Biovia Discovery studio, target proteins were generated, and water and other heteroatoms except Zn2+ were removed. The three-dimensional minimizing structures of compounds 15-20 and control drug acetazolamide at pH: 7.2 were created using the Avogadro 1.2.038 program. The target proteins hCA I (x: 35.095213, y: 16.577144, z: -18.283411) and hCA II (x: -7.312479, y: 1.358160, z: 16.219372) each have an active site coordinates file that was created with a box size of 60*60*60. Using the Discovery Studio visualizer, 2D protein-ligand interaction diagrams and 3D interactions were created.
Pharmacokinetic Studies
Substances having pharmacokinetic characteristics that enable them to persist past the end of phase I clinical trials are referred to as drug-like chemical space. The SwissADME database was used to determine the compound’s physical characteristics, such as its molecular weight, no. of H-bond acceptor, no of H-bond donor, WLOGP39, GI Absorption, BBB permeant (Blood Brain Barrier), Lipinski rule of five40, PAINS41 no. of alert. Through physicochemical property analysis, the oral bioavailability radar, sometimes referred to as the BOILED-Egg model42, was developed. This graph was created and all parameters were calculated using the SwissADME43 webserver.
Result and discussion
Scheme 1 shows the synthetic pathway that results in the synthesis of the target derivatives. Compounds 15-20 structure was verified by FTIR, 1H NMR, 13C NMR, and ESI-MS; all of the information was recorded in the supplemental data. Pyrazole with aldehyde (09-14) was made using the technique described in the publication. Pyrazole containing aldehyde, the important intermediate in this work, was made in two phases. First, hydrazones were produced when substituted acetophenones reacted with phenylhydrazine. Hydrazones are utilized right away for the following action. The second stage was treating the synthesized hydrazone with the ice-cold Vilsmeier-Haack reagent, which produced pyrazole with aldehyde molecules (09-14). which were verified by FTIR, ESI-MS, and TLC. The aldehyde group is shown by the band at about 1680 cm-1 in the FTIR graph. By reacting pyrazole with aldehyde and substituted amines with sulphonamide groups with a catalytic quantity of glacial acetic acid, Schiff bases 15-20 were synthesized. The generated compound 15-2- was found to have 1H-NMR spectra in DMSO-d6. The δ ppm suggested hydrogen present in the pyrazole ring and the δ ppm between 8.22-8.55 showed hydrogen present in the imine linkage (HC=N), which validated the production of Schiff base. A peak in 13C was found between 153.29 and 154.36 as a result of imine linkage formation. Peak in the predicted range of 144.24–145.16 (Ar-C-SO2NH2) The structure of the synthesized molecules was confirmed by mass spectral analysis, which revealed a link between the molecular ion peaks (M + 1) and the corresponding predicted molecular mass. The Supplementary Materials provide the spectra of a few typical cases.
Molecular Docking
Diverse computational methods are employed in computational research to look into and evaluate scientific issues. In computational studies, predictions are made and complicated events are understood via the use of computer simulations, algorithms, and mathematical models. A particular kind of computer research called “molecular docking” aims to forecast the orientation and binding affinity of tiny molecules, or “ligands,” within a target protein or receptor. In molecular docking, the docking process between the ligand and protein is simulated, and their compatibility and interactions are assessed. Because these investigations may be used to identify possible drug candidates based on their capacity to bind a particular target protein, they are very helpful in the discovery and design of new drugs 44,45. One of the most popular computational techniques, molecular docking, predicts the binding sites of ligands to the target and can provide important biochemical process illustrations 46–48. hCA I (PDB: 1AZM) and hCA-II (PDB: 3HS4), two human carbonic anhydrase enzymes, were the subjects of molecular docking experiments. To elucidate the tendencies noted and guarantee the precision and effectiveness of docking research, the relative selectivity and potency of our created analogues were examined. The PDB service, provided the enzymes 3D structures, which were downloaded for this purpose. The in-silico investigations of strong inhibitors were conducted using a crystal structure with excellent resolution of hCA I (2.00 Å) and hCA II (1.10 Å). Since a monomer chain is present in the crystal structures of both hCA I and hCA II, chain A was chosen for docking investigations. The outcomes of the ligands (15–20) molecular docking against the receptor binding sites are displayed in Table 1. In order to form critical interaction with zinc ions present at catalytic sites, benzenesulfonamide derivatives 17 and 19 penetrated deep into active areas of both targets. This interaction may indicate a possible method of the compound’s inhibition, which might include blocking or chelating the metal ion necessary for the activity of the enzyme implying that it could settle inside the active location. Additionally, compound 17 established pi-H interactions with TRP209, HIS94, and HIS96 amino acid residues. The chemical may generate extensive binding contacts inside the active site, which may lead to strong inhibition, as these pi-H interactions highlight. Compound 17 shows the highest binding affinity towards hCA I-17 complex, and compound 19 shows the highest binding affinity towards hCA II-19 complex. Both ligands have major H bond interactions with enzymes. Figure 2A represents the 3D binding pose of the hCA I-17 complex and Figure 2B displays the 2D docking pose of the hCA I-17 complex, while Figure 3A depicts the 3D docking pose of the hCA I-19 complex and Figure 3B depicts the 2D binding pose of the hCA II-19 complex. In the hCAI-17 complex, H bonds form between sulfonamide and THR A:199, just as in the hCAII-19 complex, H bonds form between the sulfonamide group and THR A:200. Apart from that, all the other ligands show better binding affinity than the reference drug, acetazolamide. This observation suggests that the hCAI-17 complex and hCAII-19 can interact with the receptor and stabilize the complex, as shown by their binding scores with both targets. Derivatives of benzenesulphonamides may therefore be a useful starting point for the synthesis of novel inhibitors that specifically target these enzymes.
Table 1: Binding affinity of compound 15-20 with hCA I and hCA II
|
Compound |
Binding affinity with hCA I (kcal/mol) |
Binding affinity with hCA II (kcal/mol) |
|
15 |
-8.211 |
-8.66 |
|
16 |
-9.283 |
-9.327 |
|
17 |
-9.366 |
-9.057 |
|
18 |
-8.562 |
-9.223 |
|
19 |
-8.962 |
-9.378 |
|
20 |
-8.897 |
-8.839 |
|
Acetazolamide |
-6.407 |
-6.717 |
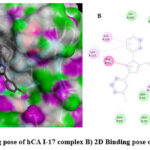 |
Figure 2: A) 3D docking pose of hCA I-17 complex B) 2D Binding pose of hCA I and 17 complex. |
 |
Figure 3: A) 3D docking pose of hCA II-19 complex B) 2D Binding pose of hCA II-19 complex. |
Pharmacokinetic Studies
Table 2: In Silico ADME for Synthesized Compound Evaluation
|
Compound |
MW |
No. of H-bond acceptor |
No of H-bond donor |
WLOGP |
GI Absorption |
BBB permeant |
Lipinski no. of violation |
PAINS no. of alert |
|
15 |
420.46 |
6 |
1 |
5.58 |
High |
No |
0 |
0 |
|
16 |
471.36 |
5 |
1 |
6.32 |
Low |
No |
0 |
0 |
|
17 |
481.36 |
5 |
1 |
5.78 |
High |
No |
0 |
0 |
|
18 |
436.91 |
5 |
1 |
5.67 |
High |
No |
0 |
0 |
|
19 |
481.36 |
5 |
1 |
5.78 |
High |
No |
0 |
0 |
|
20 |
436.91 |
5 |
1 |
5.67 |
High |
No |
0 |
0 |
It is challenging for many possible enzyme inhibitors to advance through clinical trials due to their poor pharmacokinetic characteristics. Studying the pharmacokinetics of chemicals is crucial to comprehend their fundamental characteristics. A web-based module named SwissADME was utilized to get the ADME profiles of synthesized compounds 15-20 using in silico approaches. Table 2 provides a summary of the obtained data. The gastrointestinal tract (GI) passive absorption of a drug was predicted using SwissADME. The boiled-egg plot, a straightforward approach, is included in SwissADME. An overview of the compound’s gastrointestinal absorption is shown by the egg-shaped plot. The new compound’s gastrointestinal absorption is represented by the white section of the egg, and its BBB penetration is represented by the yolk component. The figure indicates that all (except compound 16) of the synthesized compound may pass through the gastrointestinal system and did not cross the BBB, indicating adequate oral absorption. Every scaffold complied with Lipinski’s drug-likeness criteria. Furthermore, the produced hits’ promiscuity was evaluated using the Pan-assay interference compounds (PAINS alerts) filter. Every studied chemical (Table 2) exhibited druggable qualities and was without PAINS alert. Furthermore, 98.72Å topological polar surfaces and excellent gastrointestinal absorption were demonstrated by all the compounds. Given all of these factors, it is reasonable to assume that synthetic hits will prove to be effective oral therapeutic treatments for conditions resulting from carbonic anhydrase overexpression. The lipophilicity radar graph and oral bioavailability were computed using the SwissADME database, as seen in Figures 4. This graph makes use of the six physicochemical characteristics of a molecule, including size, lipophilicity, polarity, flexibility, insolubility, and insaturation. The pink zone represents the ideal physicochemical environment for oral medicine bioavailability, while the red line represents the oral drug bioavailability characteristics of the derivatives. Regarding their physicochemical makeup, all of the synthetic derivatives (15–20) are into the range delineated by the pink zone (Figures 4). For all derivatives, the instauration state is an exception.
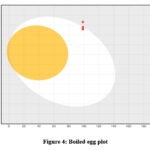 |
Figure 4: Boiled egg plot |
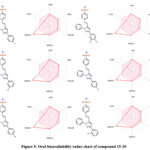 |
Figure 5: Oral bioavailability radar chart of compound 15-20. |
Conclusion
This work reports the synthesis of a novel (E)-4-((3-(substituted phenyl)-1-phenyl-1H-pyrazol-5-yl)methylene)amino)benzenesulfonamide using conventional synthetic methods. FTIR, 1H and 13C NMR, and ESI-MS were among the spectroscopic techniques used to determine the structure of this compound. A dominant diuretic agent was identified using in silico techniques. Carbonic anhydrase enzymes were selected for docking study because carbonic anhydrase inhibitors are a type of diuretic medications. The protein structures of Human Carbonic Anhydrase I (PDB ID: 1azm) and Human Carbonic Anhydrase II (PDB ID: 3hs4), which were both obtained from the Protein Data Bank, were docked using AutoDock Vina 1.2.3 software. All the synthesized ligands showed a greater binding affinity than the control medication, acetazolamide, according to the docking analysis. Of these, ligand 17 showed the most affinity for binding to hCA I protein structures, whereas ligand 19 showed the strongest affinity for binding to hCA II protein structures. Furthermore, a number of parameters were computed using the SwissADME online server in order to evaluate the ADME profile. According to the ADME analysis, every ligand had good gastrointestinal absorption and bioavailability radar, meeting the PAINS warning and Lipinski Rule of Five. But none of the ligands showed signs of being able to cross the blood-brain barrier. This in silico data provides a preliminary understanding of these compounds’ potential as effective diuretics agent. To fully verify these predictions, more in vivo research using animal models is required.
Acknowledgment
The authors gratefully acknowledge for the laboratory support provided by The HNSB. Ltd. Science College, Himatnagar
Conflict of Interests
The authors have no relevant financial or non-financial interests to disclose.
References
- Krishnamurthy, V. M.; Kaufman, G. K.; Urbach, A. R.; Gitlin, I.; Gudiksen, K. L.; Weibel, D. B.; Whitesides, G. M. ;Chem Rev 2008, 108, 946.
CrossRef - Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C. T.; De Simone, G. ;Chem Rev 2012, 112, 4421.
CrossRef - Gupta, A.; Zaheer, M. R.; Iqbal, S.; Roohi; Ahmad, A.; Alshammari, M. B. ;ACS Omega 2022, 7, 13870.
CrossRef - Goebel, C.; Trout, G. J.; Kazlauskas, R. ;Anal Chim Acta 2004, 502, 65.
CrossRef - Amendola, L.; Colamonici, C.; Mazzarino, M.; Botrè, F. ;Anal Chim Acta 2003, 475, 125.
CrossRef - Deventer, K.; Pozo, O. J.; Van Eenoo, P.; Delbeke, F. T. ;J Chromatogr A 2009, 1216, 5819.
CrossRef - Deventer, K.; Delbeke, F. T.; Roels, K.; Van Eenoo, P. ;Biomedical Chromatography 2002, 16, 529.
CrossRef - Husain, A.; Madhesia, D.; Rashid, M.; Ahmad, A.; Khan, S. A. ;J Enzyme Inhib Med Chem 2016, 31, 1682.
CrossRef - Getahun, M.; Engidawork, E. ;Ethiopian Pharmaceutical Journal 2015, 31, 35.
CrossRef - Ergena, A.; Rajeshwar, Y.; Solomon, G. ;Scientifica (Cairo) 2022, 2022, 1.
CrossRef - Hussain, T.; Ullah, S.; Alrokayan, S.; Alamery, S.; Mohammed, A. A.; Ejaz, S. A.; Aziz, M.; Iqbal, J. ;RSC Adv 2023, 13, 18461.
CrossRef - Sağlık, B. N.; Osmaniye, D.; Çevik, U. A.; Levent, S.; Çavuşoğlu, B. K.; Büyükemir, O.; Nezir, D.; Karaduman, A. B.; Özkay, Y.; Koparal, A. S.; Beydemir, Ş.; Kaplancıklı, Z. A. ;Eur J Med Chem 2020, 198, 112392.
CrossRef - Marini, A. M.; Maresca, A.; Aggarwal, M.; Orlandini, E.; Nencetti, S.; Da Settimo, F.; Salerno, S.; Simorini, F.; La Motta, C.; Taliani, S.; Nuti, E.; Scozzafava, A.; McKenna, R.; Rossello, A.; Supuran, C. T. ;J Med Chem 2012, 55, 9619.
CrossRef - Bonardi, A.; Nocentini, A.; Bua, S.; Combs, J.; Lomelino, C.; Andring, J.; Lucarini, L.; Sgambellone, S.; Masini, E.; McKenna, R.; Gratteri, P.; Supuran, C. T. ;J Med Chem 2020, 63, 7422.
CrossRef - Arslan, T.; Çelik, G.; Çelik, H.; Şentürk, M.; Yaylı, N.; Ekinci, D. ;Arch Pharm (Weinheim) 2016, 349, 741.
CrossRef - Vullo, D.; Del Prete, S.; Fisher, G. M.; Andrews, K. T.; Poulsen, S.-A.; Capasso, C.; Supuran, C. T. ;Bioorg Med Chem 2015, 23, 526.
CrossRef - Riedel, R. ;Arzneimittelforschung 1981, 31, 655.
CrossRef - Fioravanti, R.; Bolasco, A.; Manna, F.; Rossi, F.; Orallo, F.; Ortuso, F.; Alcaro, S.; Cirilli, R. ;Eur J Med Chem 2010, 45, 6135.
CrossRef - Isidro, M.; Cordido, F. ;Mini-Reviews in Medicinal Chemistry 2009, 9, 664.
CrossRef - Penning, T. D.; Talley, J. J.; Bertenshaw, S. R.; Carter, J. S.; Collins, P. W.; Docter, S.; Graneto, M. J.; Lee, L. F.; Malecha, J. W.; Miyashiro, J. M.; Rogers, R. S.; Rogier, D. J.; Yu, S. S.; Anderson, G. D.; Burton, E. G.; Cogburn, J. N.; Gregory, S. A.; Koboldt, C. M.; Perkins, W. E.; Seibert, K.; Veenhuizen, A. W.; Zhang, Y. Y.; Isakson, P. C. ;J Med Chem 1997, 40, 1347.
- Alam, R.; Wahi, D.; Singh, R.; Sinha, D.; Tandon, V.; Grover, A.; Rahisuddin ;Bioorg Chem 2016, 69, 77.
CrossRef - Jiang, D.; Zheng, X.; Shao, G.; Ling, Z.; Xu, H. ;J Agric Food Chem 2014, 62, 3577.
CrossRef - Furuya, T.; Suwa, A.; Nakano, M.; Fujioka, S.; Yasokawa, N.; Machiya, K. ;J Pestic Sci 2015, 40, 38.
CrossRef - Kaushik, D.; Khan, S. A.; Chawla, G.; Kumar, S. ;Eur J Med Chem 2010, 45, 3943.
- Abdel-Aziz, M.; Abuo-Rahma, G. E.-D. A.; Hassan, A. A. ;Eur J Med Chem 2009, 44, 3480.
CrossRef - Kumar, S.; Ceruso, M.; Tuccinardi, T.; Supuran, C. T.; Sharma, P. K. ;Bioorg Med Chem 2016, 24, 2907.
CrossRef - Nocentini, A.; Supuran, C. T. In Carbonic Anhydrases; Supuran, C. T.; Nocentini, A., Eds.; Academic Press, 2019; pp. 3–16.
CrossRef - Supuran, C. T. ;Nat Rev Drug Discov 2008, 7, 168.
CrossRef - Supuran, C. T. ;J Enzyme Inhib Med Chem 2016, 31, 345.
CrossRef - Sivaramakarthikeyan, R.; Iniyaval, S.; Saravanan, V.; Lim, W. M.; Mai, C. W.; Ramalingan, C. ;ACS Omega 2020, 5, 10089.
CrossRef - Arshad, M.; Khan, M. S.; Nami, S. A. A.; Ahmad, D. ;SN Appl Sci 2019, 1.
CrossRef - Nandurkar, Y.; Bhoye, M. R.; Maliwal, D.; Pissurlenkar, R. R. S.; Chavan, A.; Katade, S.; Mhaske, P. C. ;Eur J Med Chem 2023, 258, 115548.
CrossRef - Ashok, D.; Devulapally, M. G.; Gundu, S.; Aamate, V. K.; Chintalapally, S. ;Chem Heterocycl Compd (N Y) 2016, 52, 609.
CrossRef - Durgun, M.; Türkeş, C.; Işık, M.; Demir, Y.; Saklı, A.; Kuru, A.; Güzel, A.; Beydemir, Ş.; Akocak, S.; Osman, S. M.; AlOthman, Z.; Supuran, C. T. ;J Enzyme Inhib Med Chem 2020, 35, 950.
CrossRef - Sandhya, P. V; Muhammad Niyas, K. V; Haridas, K. R. ;Asian Journal of Chemistry 2021, 33, 1551.
CrossRef - Trott, O.; Olson, A. J. ;J Comput Chem 2009, NA.
- Eberhardt, J.; Santos-Martins, D.; Tillack, A. F.; Forli, S. ;J Chem Inf Model 2021, 61, 3891.
CrossRef - Hanwell, M. D.; Curtis, D. E.; Lonie, D. C.; Vandermeersch, T.; Zurek, E.; Hutchison, G. R. ;J Cheminform 2012, 4, 17.
CrossRef - Wildman, S. A.; Crippen, G. M. ;J Chem Inf Comput Sci 1999, 39, 868.
CrossRef - Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. ;Adv Drug Deliv Rev 2001, 46, 3.
- Baell, J. B.; Holloway, G. A. ;J Med Chem 2010, 53, 2719.
CrossRef - Daina, A.; Zoete, V. ;ChemMedChem 2016, 1117.
CrossRef - Daina, A.; Michielin, O.; Zoete, V. ;Sci Rep 2017, 7, 42717.
CrossRef - Owen, A. E.; Louis, H.; Agwamba, E. C.; Udoikono, A. D.; Manicum, A.-L. E. ;J Mol Struct 2023, 1273, 134233.
CrossRef - Babalola, I. T.; Suleiman, G. ;J Taibah Univ Med Sci 2024, 19, 175.
CrossRef - Hetényi, C.; Körtvélyesi, T.; Penke, B. ;Bioorg Med Chem 2002, 10, 1587.
CrossRef - Radwan, A. A.; Alanazi, F. K.; Raish, M. ;PLoS One 2023, 18, e0286195.
CrossRef - Anil, D. A.; Polat, M. F.; Saglamtas, R.; Tarikogullari, A. H.; Alagoz, M. A.; Gulcin, I.; Algul, O.; Burmaoglu, S. ;Comput Biol Chem 2022, 100, 107748.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


