DFT study of Lithium, Sodium and Potassium salts of Amino Acids; Global Reactivity Descriptors, Optimized parameters and Hydration Free Energy
Mahendra Bapurao Dhande1 , G Krishna Chaitanya2, Dipak Tukaram Tayade3 and Pavan Vijay Raut4*
, G Krishna Chaitanya2, Dipak Tukaram Tayade3 and Pavan Vijay Raut4*
1Department of Chemistry, HPT Arts and RYK Science College, Nashik-422005, India.
2School of Chemical Sciences, SRTM University, Nanded-431 606, India.
3Department of Chemistry, Government Vidarbha Institute of Science and Humanities,444604, Amravati, India.
4Department Of Chemistry, Smt. Radhabai Sarda Arts, Commerce and Science College Anjangaon Surji, Dist. Amravati, Pin 444705 (MH) India.
Corresponding Author E-mail: pavanraut523@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/390528
Article Received on : 03 May 2023
Article Accepted on : 16 Sep 2023
Article Published : 25 Sep 2023
Reviewed by: Dr. Sreehari Sastry
Second Review by: Dr. Mohd. Mohsin
Final Approval by: Dr. Charanjit Kaur
A promising solvent for removing CO2 from flue gases after combustion is aqueous solutions of amino acid salts of alkali metals. Their computational work would be extremely significant in this area. Amino acid salts of alkali metals are being explored in aqueous solutions for post-combustion CO2 capture from flue gases. In this regard, their computational study would be of the utmost importance. The hydration free energies, total dipole moment, HOMO/LUMO band gap energy, C=O vibration of the –COOH group, bond lengths and bond angles for lithium, sodium and potassium cysteinate and prolinate were computed in the current work using the Gaussian 09 program. Study shows that the hydration free energy for potassium saltsisless than that of corresponding lithium and sodium salts. From result it could be stated that the change of alkali metal(Na/ K)in amino acid saltsare changing the physical structural and vibrational characteristics of amino acid salts. This study would be helpful for their evaluation as a CO2 capturing agent.
KEYWORDS:CO2; DFT; Hydration free energies; total dipole moment
Download this article as:| Copy the following to cite this article: Dhande M. B, Chaitanya G .K, Tayade D. T, Raut P. V. DFT study of Lithium, Sodium and Potassium salts of Amino Acids; Global Reactivity Descriptors, Optimized parameters and Hydration Free Energy. Orient J Chem 2023;39(5). |
| Copy the following to cite this URL: Dhande M. B, Chaitanya G .K, Tayade D. T, Raut P. V. DFT study of Lithium, Sodium and Potassium salts of Amino Acids; Global Reactivity Descriptors, Optimized parameters and Hydration Free Energy. Orient J Chem 2023;39(5). Available from: https://bit.ly/455KAil |
Introduction
Major greenhouse gas carbon dioxide (CO2) is accountable for the global warming that has been observed over the past few decades, as well as for the worries about relatedclimate change and its potential impact on humanity. Consequently, the removal of CO2 from a process gas is a crucial step in many industrial processes. Prior to the flue gas being released into the atmosphere, CO2 is separated from the flue gas using CO2 capture methods. Many researchers have examined the responses of the amino acid salt solution with CO21-3, which is developing into a CO2 capture absorbent. Their physical characteristics, optimum geometrical parameters, and molecular characteristics (Global Reactivity Descriptors) may be useful for assessing and characterizing them as a CO2 collecting absorbent and for other industrial uses.
For lithium cysteinate, sodium cysteinate and potassium cysteinate, lithium prolinate, sodium prolinate and potassium prolinate such properties have not yet been reported in the open literature. Thus, in the present work, we presented new data on the optimized geometrical parameters like bond length LO-M and bond angle O-C=O of –COOM group(M=Na/k), molecular properties including chemical hardness(η), softness (S), chemical potential (µ) and electronegativity (χ) and physical parameters like hydration free energies(HFE), total dipole moment, HOMO/LUMO band gap energy, C=O vibration of COOH group, bond length and bond angles inlithium cysteinate, sodium cysteinate and potassium cysteinate, lithium prolinate, sodium prolinate and potassium prolinate.
 |
Figure 1: Structures of Li-cyst, Na-cyst, K-cyst, Li-pro, Na-pro and K-pro. |
Experimental Section
Computational Work
Computational work was performed using Gaussian 09 software. Lithium cysteinate(Li-cyst), sodium cysteinate(Na-cyst), and potassium cysteinate(K-cyst), as well as lithium prolinate(Li-pro), sodium prolinate(Na-pro) and potassium prolinate(K-pro), hydration free energies and other optimal parameters were estimated in the current work in both the gas phase and the aqueous phase. The geometry of amino acids salts (AAS) was fully optimised to calculate the various parameters and hydration free energy and the frequencies were calculated by using the density functional theory (DFT) B3LYP method at the 6-31++ G (d, p) basic set4 using the PCM (Polarizable Continuum Model) solvation Model.
Results and Discussion
Optimized geometrical parameters (bond length (L) in Å and angle (A) in Deg.)
For Li-cyst, Na-cyst, K-cyst, Li-pro, Na-pro, and K-pro, the optimized geometrical parameters, suchas bondlength LO-M, and O-C=O of the -COOM group (M=Na/k), were computed5and are presented in Table 1. The O-M bond length increases from Li-AAS to Na-AAS and from Na-AAS to K-AAS. This is most likely attributed to an increase in the metal’s size in gas and aqueous phases. The O-M bond length increases for M= Li, Na and K when transition from the gas phase to the aqueous phase. From Li-AAS to Na-AAS and from Na-AAS to K-AAS, the O-C=O bond angles increases. This O-C=O bond angel increase is also observed from gas to aqueous phase. This pattern is seen across all examined AAS. In Li-cyst, Na-cyst, K-cyst, the O-M bond length and O-C=O bond angles are less than that of corresponding Li-pro, Na-pro and K-pro salts.
Li-AAS (O-M) L< Na-AAS (O-M) L< K-AAS (O-M)L
pro-AAS(O-M) L < cyst-AAS (O-M) L
Li-AAS (O-C=O) A< Na-AAS(O-C=O) A< K-AAS(O-C=O) A
pro-AAS (O-C=O) A< cyst-AAS (O-C=O) A
Molecular properties (Global Reactivity Descriptors)
The eigenvakue of the highest-occupied molecular orbital(HOMO), lowest occupied molecular orbital(LUMO),electronegativity, HOMO-LUMO gap and chemical hardness are the most well-known of these parameters (Global Reactivity Descriptors) that are based on the DFT6,7. The 1930s-era Koopmans theorem8 provides a different method for predicting the ionization energy and electron affinities of chemical species and serves as a link between conceptual density functional theory and molecular orbital theory. This theory states thatthe ionization energy and electron affinity of a molecule are roughly similar to the negative values of those orbitals’ HOMO and LUMO energies. The energy difference between the HOMO and LUMO states is crucial aspect in defining the molecular electrical transport capacities9. Global chemical reactivity descriptors10, such as chemical hardness(𝜼), chemical potential(μ), softness(S) and electronegativity(χ) of molecules have been defined using the HOMO and LUMO energy values for a molecule11–13. Using Koopmans’ theorem14 and the HOMO and LUMO energy values, chemical hardness, chemical potential, electronegativity, and softnessof closed-shell molecules are defined as follows,

where, I denotes the ionisation potential and Ais the electron affinity. HOMO and LUMO orbital energies can be used to express I and A as I = -E HOMO and A = -E LUMO15.
Resistance to changes in electronic configurationof a chemical speciesis evaluated by their chemical hardness16–18. It also has some important applications in topic like complex stability, chemical reactivity19, solubility of molecules and in estimation of formed product of the reaction20. Calculated values of ionization product, electron affinity, hardness, potential and softness for Li-cyst, Na-cyst, K-cyst, Li-pro, Na-pro and K-pro, are presented in Table 2.Hardness and softness can also have an impact on a molecule’s stability21. In thegaseous and aqueous phases, the chemical hardness and electronegativity values of K-AAS are marginally lower than those of corresponding Na-AAS and for Na-AAS, they are marginally lower than those of corresponding Li-AAS. The only exception is for the value for Na-AAS in the gaseous phase. Cyst-salt values are higher than pro-salt readings. All of the examined AAS experience an increase in and a decrease in electronegativity as they transition from a gaseous to an aqueous solution.
It is clear from the information in Tables 2 that the η value for Li-cyst is maximum in both the aqueous phase and the gas phase.
The combined molecular properties data for the studied structures Na-gly, Na-ala, Na-val, Na-leu, K-gly, K-ala, K-val, and K-leu indicate that:
Chemical hardness and chemical potential values are higher in aqueous phase as compare to gas phase; whereas, softness and electronegativity show lower values for all studied structures.
Li-AAS, Na-AAS, and K-AAS have been shown to have the following observed trends for η and χ.
cyst-salt < η pro-salt
Li-salt >η Na-salt > η K-salt
χ Li-salt >χ Na-salt >χ K-salt -aqueous phase
 |
Figure 2: Optimized structures for Li-cyst, Na-cyst, K-cyst, Li-pro, Na-pro and K-pro. |
Physical parameters viz.total dipole moment(TDM) , HOMO-LUMOband gap energy(ΔE) and the C=Ovibration in carboxylic group.
In addition, the computed ΔE, TDM values, and C=O vibration of the carboxylic group of the investigated AAS in both gas and water phase. The two physical parameters TDM and E measure a given compound’s reactivity22,23 and stability24 with the molecules around it. Reactive chemicals are indicated by a high TDM and a low ΔE25. From Li-AAS to Na-AAS and from Na-AAS to K-AAS, the total dipole moment increases while the band gap energy marginally lowers for both amino acids.
(ΔE) Li-salt >(ΔE) Na-salt > (ΔE) K-salt,
(TDM) Li-salt <(TDM) Na-salt < (TDM) K-salt,
Aqueous phase (TDM and ΔE) > Gas phase (TDM and ΔE)
Trend of total dipole moment(TDM) in both phase for AAS as shown below
(TDM) pro-salts>(TDM) cyst-salts
Trend of HOMO/LUMO band gap energy(ΔE)in both phase for AAS as
(ΔE)pro-salts <(ΔE)cyst-salts
The aforementioned trend clearly indicates that for the examined AAS, the TDM and ΔE values are larger in aqueous solutions compared to gas phase. Figures 3 and 4 present a graphic comparison of the ΔE and TDM of Li-AAS, Na-AAS, and K-AAS, respectively. In both phases, it was discovered that K-pro had the lowest ΔE values, indicating that it was highly reactive, whereas Li-cyst had the highest ΔE values, indicating that it was the least reactive.
Due to an increase in the ionic radius of the metal ion, the C=O vibrational characteristic band of the carboxyl group was significantly shifted toward higher wavenumber from Li-AAS to Na-AAS and from Na-AAS to K-AAS. Observations show that K-pro has the highest and Li-pro has the lowest values.
Calculated Hydration Free Energies for all studied AAS molecules
Understanding the free energy of solvation, which is the energy required for a molecule to go from gas phase to solution, is essential for understanding how chemistry works in solutions. When water is the solvent, the term “solvation free energy” is often used to describe the hydration process. The hydration free energy26 determines how stable an ion is in its hydrated state compared to how stable it is in its anhydrate condition. Hydration free energy was calculated using the DFT B3LYP method at the basis set of 6-31++G (d, p) (HFE),which is the work required to transfer a molecule from the gas phase into the solution phase. To calculate HFE, use the following equation.
Δ G0Hydr = ΔG0S – ΔG0g
where ΔG0g and ΔG0Srepresents the standard free energy of solute in the gas phase and in the solvent respectively, and G0Hydr refers as HFE. Table 4 and Figure 5 compare the HFE of the interested AAS. HFE for all AAS under investigation is negative, indicating a strong solute-solvent interaction. Na-salts have greater HFE values than the corresponding Li- and K-salts for amino acids. Among the examined AAS, Na-cyst was shown to have the highest HFE values.
The observed trend for the HFE values for all studied AAS is
(HFE) K-AAS<(HFE) Li-AAS<(HFE) Na-AAS
Table 1: Optimized geometrical parameters: (bond length (L) in Å and bond angle (A) in Deg.) for all studied AAS molecules obtained at B3LYP/6-311+G(d,p) level.
|
Optimized geometrical parameters |
Li-cyst |
Na-cyst |
K-cyst |
|
|
Gas Phase |
||
|
L(O-M) |
1.8610 |
2.1968 |
2.5537 |
|
A(O-C=O) |
121.29 |
123.73 |
124.8 |
|
|
Aqueous Phase |
||
|
L(O-M) |
2.0518 |
2.3561 |
2.7667 |
|
A(O-C=O) |
122.71 |
124.33 |
125.27 |
|
|
Li-pro |
Na-pro |
K-pro |
|
|
Gas Phase |
||
|
L(O-M) |
1.8525 |
2.1956 |
2.5497 |
|
A(O-C=O) |
120.92 |
123.43 |
124.47 |
|
|
Aqueous Phase |
||
|
L(O-M) |
2.0251 |
2.3559 |
2.7597 |
|
A(O-C=O) |
122.39 |
124.12 |
125.04 |
Table 2: Molecular propertiesviz.chemical hardnes (η), softness(S), chemical potential (m) and electronegativity(c).
|
Molecular Properties |
Li-cyst |
Na-cyst |
K-cyst |
|
|
Gas Phase |
||
|
I |
6.2706 |
5.9792 |
5.7648 |
|
A |
1.1023 |
1.6183 |
1.4294 |
|
chemical hardness(η) |
2.5842 |
2.1805 |
2.1677 |
|
softness (S) |
0.3870 |
0.4586 |
0.4613 |
|
chemical potential(m) |
-3.6865 |
-3.7988 |
-3.5971 |
|
electronegativity(c) |
3.6865 |
3.7988 |
3.5971 |
|
|
Aqueous Phase |
||
|
I |
6.6622 |
6.6266 |
6.5950 |
|
A |
0.4060 |
0.4169 |
0.4229 |
|
chemical hardness(η) |
3.1281 |
3.1049 |
3.0861 |
|
softness (S) |
0.3197 |
0.3221 |
0.3240 |
|
chemical potential(m) |
-3.5341 |
-3.5218 |
-3.5090 |
|
electronegativity(c) |
3.5341 |
3.5218 |
3.5090 |
|
|
Li-pro |
Na-pro |
K-pro |
|
|
|
Gas Phase |
|
|
I |
5.6461 |
5.3313 |
5.0690 |
|
A |
1.1083 |
1.6117 |
1.4278 |
|
chemical hardness(η) |
2.2689 |
1.8598 |
1.8206 |
Table 3: Physical parameters for all studied AAS viz. ΔE, TDM and the C=O vibration in carboxyl group.
|
Molecular Properties |
Li-cyst |
Na-cyst |
K-cyst |
|
|
|
Gas Phase |
|
|
ΔE (eV) |
5.1683 |
4.3609 |
4.3354 |
|
TDM (D) |
3.6075 |
5.8056 |
7.8980 |
|
C=O vibration |
1580.3700 |
1592.5600 |
1601.5300 |
|
|
Aqueous Phase |
||
|
ΔE (eV) |
6.2562 |
6.2097 |
6.1721 |
|
TDM (D) |
5.1733 |
7.2182 |
9.5197 |
|
C=O vibration |
1573.3900 |
1577.5800 |
1582.1400 |
|
|
Li-pro |
Na-pro |
K-pro |
|
|
Gas Phase |
||
|
ΔE (eV) |
4.5378 |
3.7196 |
3.6412 |
|
TDM (D) |
4.3832 |
6.7758 |
7.3595 |
|
C=O vibration |
1567.7000 |
1587.5300 |
1600.4900 |
|
|
Aqueous Phase |
||
|
ΔE (eV) |
5.8722 |
5.7210 |
5.6277 |
|
TDM (D) |
6.4383 |
8.5160 |
8.9823 |
|
C=O vibration |
1571.4000 |
1579.2400 |
1584.3100 |
Table 4: Calculated Hydration Free Energies for all studied AAS molecules.
|
Molecular Properties |
Li-cyst |
Na-cyst |
K-cyst |
|
Hydration Free Energy (Kcal/mol) |
-33.08 |
-33.16 |
-29.26 |
|
|
Li-pro |
Na-pro |
K-pro |
|
Hydration Free Energy (Kcal/mol) |
-30.66 |
-31.12 |
-27.52 |
 |
Figure 3: Comparison of DE of Li-cyst, Na-cyst, K-cyst, Li-pro, Na-pro and K-pro. |
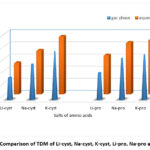 |
Figure 4: Comparison of TDM of Li-cyst, Na-cyst, K-cyst, Li-pro, Na-pro and K-pro. |
 |
Figure 5: Comparison of Hydration Free Energies of Li-cyst, Na-cyst, K-cyst, Li-pro, Na-pro and K-pro. |
Conclusion
As the O-M bond length increases from Li-AAS to Na-AAS and from Na-AAS to K-AAS with a change in the (C-O=C) bond angle, the geometry of the studied amino acid salt changes. All of the examined AAS experience an increase in η andχas they transition from a gaseous to an aqueous solution. It is known that both the aqueous phase and the gas phase of Li-cyst have the greatest η values. As a result, we can say that it is the least reactive.
Both the TDM and ΔE varies as a result of increased size of metal and the alkyl part of studied amino acid. Lower ΔE and higherTDM values are the indicative of greater reactivity of K-AAS compare to Na-AAS. ΔE values of cyst-salt are higher than that of pro-salt. K-cyst has found to be highest dipole moment among all studied salts in both phases. The change in the geometrical parameters shiftsC=O characteristic band toward lower wavenumbers.Negative value of the HFE of AAS suggests strong interaction between AAS and water. The hydration free energy is highest for Na-AAS compare to Li-AAS and K-AAS.
Acknowledgement
MBD thanks the Director of the Government Vidarbha Institute of Science and Humanities in Amravati and the Principal of the HPT Arts and RYK Science College in Nashik for providing research facilities and their kind cooperation.
Conflicts of Interest
Authors declares that there is no conflict of interest.
References
- Zhang, Z., Rao, S., Han, Y., Pang, R., and Ho, W. W. (2021). CO2-selective membranes containing amino acid salts for CO2/N2 separation. Journal of Membrane Science, 638, 119696.
CrossRef - Liu, M., and Gadikota, G. (2020). Single-step, low temperature and integrated CO2 capture and conversion using sodium glycinate to produce calcium carbonate. Fuel, 275, 117887.
CrossRef - Soroush, E., Mesbah, M., Hajilary, N., and Rezakazemi, M. (2019). ANFIS modeling for prediction of CO2 solubility in potassium and sodium based amino acid Salt solutions. Journal of Environmental Chemical Engineering, 7(1), 102925.
CrossRef - M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
CrossRef - Tayade, K., Bondhopadhyay, B., Basu, A., Chaitanya, G. K., Sahoo, S. K., Singh, N., & Kuwar, A. (2014). A novel urea-linked dipodal naphthalene-based fluorescent sensor for Hg (II) and its application in live cell imaging. Talanta, 122, 16-22.
CrossRef - Kaya, S., Tüzün, B., Kaya, C., & Obot, I. B. (2016). Determination of corrosion inhibition effects of amino acids: quantum chemical and molecular dynamic simulation study. Journal of the Taiwan Institute of Chemical Engineers, 58, 528-535.
CrossRef - Valaboju, A., Gunturu, K. C., Kotamarthi, B., Joly, D., & Hissler, M. (2017). DFT study of Host-Dopant systems of DPVBi with Organophosphorus π-Conjugated materials. Computational and Theoretical Chemistry, 1113, 61-71.
CrossRef - Koopmans, T. (1933). Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica, 1, 104-113.
CrossRef - Zhang, G., & Musgrave, C. B. (2007). Comparison of DFT methods for molecular orbital eigenvalue calculations. The journal of physical chemistry A, 111(8), 1554-1561.
CrossRef - Erdogan, Ş., Safi, Z. S., Kaya, S., Işın, D. O., Guo, L., & Kaya, C. (2017). A computational study on corrosion inhibition performances of novel quinoline derivatives against the corrosion of iron. Journal of Molecular Structure, 1134, 751-761.
CrossRef - Miar, M., Shiroudi, A., Pourshamsian, K., Oliaey, A. R., & Hatamjafari, F. (2021). Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo [d] thiazole-2 (3 H)-imine and its para-substituted derivatives: Solvent and substituent effects. Journal of Chemical Research, 45(1-2), 147-158.
CrossRef - Choudhary, V., Bhatt, A., Dash, D., & Sharma, N. (2019). DFT calculations on molecular structures, HOMO–LUMO study, reactivity descriptors and spectral analyses of newly synthesized diorganotin (IV) 2‐chloridophenylacetohydroxamate complexes. Journal of computational chemistry, 40(27), 2354-2363.
CrossRef - Khan, S. A., Rizwan, K., Shahid, S., Noamaan, M. A., Rasheed, T., & Amjad, H. (2020). Synthesis, DFT, computational exploration of chemical reactivity, molecular docking studies of novel formazan metal complexes and their biological applications. Applied Organometallic Chemistry, 34(3), e5444.
CrossRef - Pearson, R. G. (1997). Chemical hardness (Vol. 10, No. 3527606173). Weinheim: Wiley-VCH.
CrossRef - Lucio, F. N. M., da Silva, J. E., Marinho, E. M., Mendes, F. R. D. S., Marinho, M. M., & Marinho, E. S. (2020). Methylcytisine alcaloid potentially active against dengue virus: a molecular docking study and electronic structural characterization. Int J Res, 8(1), 221-36.
CrossRef - Ibrahim, M. (2009). Molecular modeling and FTIR study for K, Na, Ca and Mg coordination with organic acid. Journal of Computational and Theoretical Nanoscience, 6(3), 682-685.
CrossRef - Abad, N., Hajji, M., Ramli, Y., Belkhiria, M., Moftah H. Elmgirhi, S., A. Habib, M., … & Essassi, E. M. (2020). A newly synthesized nitrogen‐rich derivative of bicyclic quinoxaline—Structural and conceptual DFT reactivity study. Journal of Physical Organic Chemistry, 33(6), e4055.
CrossRef - Chakraborty, D., & Chattaraj, P. K. (2021). Conceptual density functional theory based electronic structure principles. Chemical Science, 12(18), 6264-6279.
CrossRef - Mebi, C. A. (2011). DFT study on structure, electronic properties, and reactivity of cis-isomers of [(NC5H4-S) 2Fe (CO) 2]. Journal of Chemical Sciences, 123(5), 727-731.
CrossRef - Makov, G. (1995). Chemical hardness in density functional theory. The Journal of Physical Chemistry, 99(23), 9337-9339.
CrossRef - Dheivamalar, S., & Banu, K. B. (2019). A DFT study on functionalization of acrolein on Ni-doped (ZnO) 6 nanocluster in dye-sensitized solar cells. Heliyon, 5(12), e02903.
CrossRef - Ibrahim, M., & Mahmoud, A. A. (2009). Computational notes on the reactivity of some functional groups. Journal of Computational and Theoretical Nanoscience, 6(7), 1523-1526.
CrossRef - Ezzat, H. A., Hegazy, M. A., Nada, N. A., & Ibrahim, M. A. (2019). Effect of nano metal oxides on the electronic properties of cellulose, chitosan and sodium alginate. Biointerface Res. Appl. Chem, 9, 4143-4149.
CrossRef - Singh, J. S. (2020). IR and Raman spectra with Gaussian-09 molecular analysis of some other parameters and vibrational spectra of 5-fluoro-uracil. Research on Chemical Intermediates, 46(5), 2457-2479.
CrossRef - Miar, M., Shiroudi, A., Pourshamsian, K., Oliaey, A. R., & Hatamjafari, F. (2021). Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo [d] thiazole-2 (3 H)-imine and its para-substituted derivatives: Solvent and substituent effects. Journal of Chemical Research, 45(1-2), 147-158.
CrossRef - Tansel, B. (2012). Significance of thermodynamic and physical characteristics on permeation of ions during membrane separation: Hydrated radius, hydration free energy and viscous effects. Separation and purification technology, 86, 119-126.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


