Highly Efficient Chemoselective Synthesis of 2-Aryl-1-arylmethyl-1H-Benzimidazoles by Using TiCp2Cl2 Catalyst
Samiran Halder1 and Arup Datta2*
1Department of Chemistry, Charuchandra College, 22, Lake Road, Kolkata-700029, India.
2Department of Chemistry, Shibpur Dinobundhoo Institution (College), 412/1 G.T Road (South), Shibpur, Howrah, India.
Corresponding Author E-mail: arupdattadb@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/360623
Article Received on : 17-08-2020
Article Accepted on :
Article Published : 30 Dec 2020
One pot highly efficient chemoselective N-arylmethyl benzimidazole has been prepared by using catalyst TiCp2Cl2 in tetrahydrofuran solvent smoothly. This method is very much effective for the condensation between different aldehydes and various ortho-phenylenediamine to synthesize N-substituted benzimidazole. Clean reaction procedure, quick reaction, easy purification and high yield of the product are some advantages of this protocol. All the target molecules were identified by spectroscopic analysis and also verified melting points from literature value.
KEYWORDS:Bis(cyclopentadienyl) titanium(IV) dichloride; Chemoselective synthesis: N-arylmethyl Benzimidazole; Ortho-Phenylenediamines
Download this article as:| Copy the following to cite this article: Halder S, Datta A. Highly Efficient Chemoselective Synthesis of 2-Aryl-1-arylmethyl-1H-Benzimidazoles by Using TiCp2Cl2 Catalyst. Orient J Chem 2020;36(6). |
| Copy the following to cite this URL: Halder S, Datta A. Highly Efficient Chemoselective Synthesis of 2-Aryl-1-arylmethyl-1H-Benzimidazoles by Using TiCp2Cl2 Catalyst. Orient J Chem 2020;36(6). Available from: https://bit.ly/3lpqUil |
Introduction
N-arylmethyl benzimidazoles have tremendous application in the biochemical and medicinal field.1 1,2-Disubstituted Substituted benzimidazole derivatives are used as anthelmintic agents in veterinarian medicine and treatment of antihistamineics.2 They have remarkable applications in several diseases such as antiviral agent against several viruses such as HIV,3 antimicrobial agents,4 antititimor,5 influenza,6 antiprotozoal 7 and anti-inflammatory agents.8-10 The traditional procedure of 1,2-disubstitued benzimidazoles have been prepared by using an ortho-diamines and derivatives of carboxyl acids such as amidates, orthoesters and nitriles under hot reaction method.11,12 The second route is the condensation between aldehyde and ortho-phenylenediamines in presence of various Lewis acid catalysts.13,14 There are some different methods for preparation of disubstituted benzimidazoles by applying resin supported solid-phase, 15 microwave assisted reaction16 and green solvent mediated synthesis such as water or ionic liquids.17
However many of these methodologies suffer disadvantages like low yields, no easy accessibility of the materials, harsh and lengthy time, extremely elevated temperature, additive oxidising catalyst, column chromatography for purification of products and formation of considerable amount of byproduct.18-20 Still now it is very much crucial to generate a simple and efficient pathway for the production of 1,2-disubstitued benzimidazoles. An efficient method was published to synthesis related heterocyclic compound with excellent yield by using poisonous Ti(OBu)4 catalyst under mild conditions.21,22 Another methodology was reported to synthesize alkynes by using TiCp2Me2.23 So Ti metal complex provides enormous interest to the researcher to develop new synthesis through the improvement of new methodology for the preparation of heterocyclic compounds.
An effective and generalised easy method has been reported for the generation of N-Arylmethyl benzimidazoles 3 by condensation between aromatic aldehydes and ortho-phenylenediamines using TiCp2Cl2 [Bis(cyclopentadienyl)titanium(IV) dichloride] as a catalyst (Scheme 1) in THF solvent.
Results and Discussions
Methodological organic synthesis always gets attractive interest to the chemist. A new methodology has been developed for the formation of N-substituted benzimidazole in organic solvent. Initially optimization of the reaction conditions have been set up by using p-chlorobenzaldehyde (2a, 2 mmol) and ortho-phenylenediamine (1a, 1.2 mmol) in presence of catalyst under different conditions to examine the influence of catalytic activity TiCp2Cl2 (Scheme 1) in this work. In THF solvent at 90 OC the reaction was stirred with different amount of catalyst reported in Table 1. It was found that without any catalyst very low yield (20 mol%) of the desired compound 3a was isolated and it was confirmed by TLC experiment. Then 5 mol % of catalyst was loaded but improved yield of the product 3a was only 40%. It was noticed that the amount of product was increasing as the amount of catalyst increased. In presence 20 mol% of catalyst 90% product was found and the quantity of the product did not change with increasing the time and increasing the amount of catalyst (Entry 7, Table 1).At60 OC only 60% yield was observed (Entry 6, table 1) even the reaction was continued for 10 hrs. No improved yield wasisolated even the reaction was carried out at 110 OC in THF solvent (Entry 10, Table 1). So finally conclusion has been made that 20 mol% of the TiCp2Cl2 was appropriate to develop the large yield of the desired product 3a. From the graphical representation (Figure 1) it is clear that there is no increasing of the of the yield of the product is observed in presence of more than 20 mol% of the catalyst at 90 oC (Entries 7 and 9, Table 1).
 |
Scheme 1: Synthesis of N-arylmethylbenzimidazoles in presence of TiCp2Cl2 Click here to View scheme |
Table 1: Examination of the amount of catalyst required in the reaction
|
Entry
|
TiCp2Cl2 (mol%) |
aConditions |
Time (h) |
bYield (%) |
|
1 |
0 |
90 oC, THF |
10 |
20 |
|
2 |
05 |
90 oC, THF |
10 |
40 |
|
3 |
10 |
90 oC, THF |
10 |
50 |
|
4 |
15 |
90 oC, THF |
10 |
70 |
|
5 |
20 |
90 oC, THF |
6 |
90 |
|
6 |
20 |
60 oC, THF |
10 |
60 |
|
7 |
25 |
90 oC, THF |
6 |
90 |
|
8 |
25 |
60 oC, THF |
10 |
60 |
|
9 |
30 |
90 oC, THF |
6 |
92 |
|
10 |
20 |
110 oC,THF |
6 |
88 |
|
Conditions: p-chlorobenzaldehyde (2 mmol) and ortho-phenylenediamine (1.2 mmol),THF (5mL) bIsolated Yield. |
||||
 |
Figure 1: Graphical representation of the different mol% of catalyst and % yield of the product at 90 oC (3a) |
After the initialization of the mol% of the catalyst then next experiment was designed to determine the appropriate solvent in which the reaction occurs smoothly shown in Table 2. For this purpose 20 mol% of TiCp2Cl2 were used in different solvents at 90 oC for 6 hrs. It was found that in aqueous alcoholic medium and aqueous medium the yield was 60% and 45% respectively (Entries 5 and 6, Table 2) and low yield can be attributed because of the loss of homogeneity in the reaction medium. In neat condition the yield was only 40% because the reaction medium was heterogeneous (Entry 8, Table 2). In other solvents like acetonitrile the yield was 70% (Entry 3, Table 2) and 65% yield (Entry 4, Table 2) was observed in presence of ethanol solvent. In presence of THF solvent chemo selectivity of the product raised and desired product 3a was obtained selectively as a large amount 90% (Entry 2, Table 2).
Table 2: Choice of the solvent for the formation of compound 3a
|
Entry |
Solvent (5 mL) |
aConditions |
Time (h) |
bYield (%) |
|
1 |
Toluene |
TiCp2Cl2 (20 mol%), (90 oC) |
6 |
65 |
|
2 |
THF |
TiCp2Cl2 (20 mol%), (90 oC) |
6 |
90 |
|
3 |
MeCN |
TiCp2Cl2 (20 mol%), (reflux) |
6 |
70 |
|
4 |
EtOH |
TiCp2Cl2 (20 mol%), (90 oC) |
6 |
65 |
|
5 |
EtOH+H2O (1:1) |
TiCp2Cl2 (20 mol%), (90 oC) |
6 |
60 |
|
6 |
H2O |
TiCp2Cl2 (20 mol%), (90 oC) |
6 |
45 |
|
7 |
CH2Cl2 |
TiCp2Cl2 (20 mol%), (reflux) |
6 |
60 |
|
8 |
Neat |
TiCp2Cl2 (20 mol%), (90 oC) |
6 |
40 |
|
Conditions: p-chlorobenzaldehyde (2 mmol) and o-diamine (1.2 mmol), bIsolated Yield. |
||||
So by combining the above two experiments we can say that our dream came true to develop the methodology at 90 oC in presence of 20 mol% of TiCp2Cl2 catalyst in THF solvent with better yield of the desired product 3a. Then we had applied this successful methodology to generate different N-arylmethyl benzimidazoles to generalise the scheme reported in Table 3.
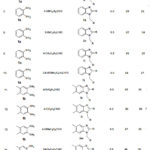 |
Table 3: Synthesis of 2-Aryl-1-arylmethyl-1H-benzimidazoles |
This condensation reaction became successful to generate the desired product 3a in good yield under this reaction condition with different aromatic aldehydes and various ortho-phenylenediamine. Although the reaction was catalyzed by TiCp2Cl2 but % yield and time of the completion of the reaction the products depend on the reactivity of the aldehyde group in aromatic aldehyde moiety depicted in Table 3. Usually electron withdrawing group at the meta and Para position provided excellent yield of the products rather than the electron pushing group at the same position present in aromatic aldehydes and that’s why excellent yields were observed in presence of functional group Cl, Br, CN and F (Entries 1, 2, 3 and 19, Table 3) with respect to electron donating methoxy and methyl group (Entries 4 and 9, Table 3). In case of para-nitrobenzaldehyde comparatively higher yield was found and less time was required to end the reaction. Benzaldehyde gave low yield of the product may be because of aerial oxidation of the substrate during the reaction. Para-hydroxybenzaldehyde also gave good yield of the product and as well as 3,4-dimethoxybenzaldehyde gave the same result (entry 7, Table 3) but in both cases requisite larger time to finish the reaction (Entry 10, Table 3). Even using dimethyl and dichloro ortho-phenylenediamine better yield of the product was achieved with different aldehydes. Not only aromatic aldehyde but this methodology was equally effective in the case of hetero aromatic aldehyde also (Entry 20, Table 3). So this methodology was very much regular in each reaction and more effective than other published paper26.
Plausible reaction mechanism for the generation of N-arylmethyl benzimidazole
Here Ti metal complex act as a strong Lewis acid and it increases the electrophilicity of the aldehyde carbonyl carbon by coordination21 with oxygen atom and diimine formation took place immediately reported in previous published paper29 and then cyclisation followed by H+ ion transfer generates the desired products with good yield shown in mechanism (Scheme 2). It has been proved from the above analysis that our methodology is very general for the synthesis of N-disubstituted benzimidazoles with wide variations in both the ortho-phenylenediamine and the aromatic aldehyde in THF solvent at 90 oC without any formation of the side product 4 (Scheme 1).
 |
Scheme 2: Detailed mechanism for N-arylmethyl benzimidazole synthesis Click here to View Scheme |
Experimental
All chemicals and solvents are used AR grade. All aromatic aldehydes and o-phenylenediamine were purchased from Spectrochem, Pvt. Ltd. Mumbai, India. TiCp2Cl2 was purched from Sigma Aldrich.FT-IR spectra and NMR spectra were recorded on Perkin-Elmer spectrometer and Bruker 300 MHz spectrometer respectively. For melting points measurement a Gallenkamp electrothermal apparatus was used.
General methodology for the synthesis of N-arylmethylbenzimidazole
In a round-bottomed flask substituted benzaldehyde (2, 2 mmol) and ortho-phenylenediamine (1, 1.2 mmol) (Scheme 1) were thoroughly mixed and dissolved in 5 mL tetrahydrofuran solvent. Then 20 mol% TiCp2Cl2 catalyst was added and stirred the mixture vigorously. After a while the reaction was checked by silica coated TLC plate in presence of ethyl acetate and petroleum ether as solvent. The reaction completed within 5.0-6.5 hrs. Then the mixture was poured in 5 mL water and kept the reaction mixture in a refrigerator for overnight. Separated crude product was crystallized directly by aqueous EtOH. Character analysis of product (3a-t) was compared by FT-IR, 1H NMR and 13C NMR data with known compounds.
Selected characterization data for synthesized compounds
2-(3, 4-Dimethoxyphenyl)-1-(3′, 4′-dimethoxyphenylmethyl)-1H-benzimidazole (3j)
White M.P. 170-172 ºC (Aqueous EtOH), FT-IR (KBr, cm-1): 2918, 2376, 1507, 1451, 1248, 1135 and 102, 1H NMR (300 MHz, CDCl3) δ: 7.86 (d, 1H), 7.33-7.28 (m, 2H), 7.24-7.22 (m, 3H), 6.91 (d, 1H), 6.79 (d, 1H), 6.65-6.61 (m, 2H), 5.38 (s, 2H), 3.90 (s, 3H), 3.84 (s, 3H), 3.77 (s, 3H), 3.76 (s, 3H), 13C NMR (75MHz, CDCl3) δ: 153.9, 150.3, 149.3, 148.9, 148.4, 142.7, 136.1, 128.9, 122.7, 122.5, 122.3, 121.7, 119.5, 118.0, 112.2, 111.4, 110.9, 110.1, 108.9, 55.8, 55.7, 55.6, 47.9, Anal. Calculated for C24H24N2O4(%): C, 71.27; H, 5.98; N, 6.93; Found: C, 71.15; H, 5.72; N, 6.69 %.
5, 6-Dichloro-2-(4-methoxyphenyl)-1-(4′-methoxyphenylmethyl)-1H-benzimidazole (3q)
White, M.p. 167-168 ºC (Aqueous EtOH), FT-IR (KBr, cm-1): 3434, 2378, 1611, 1515, 1459, 1299, 1250, 1176 and 773, 1H NMR (300 MHz, CDCl3) δ: 7.90 (s, 1H), 7.62 (dd, 2H), 7.27 (dd, 2H), 7.01-6.70 (m, 3H), 6.88 (dd, 2H), 5.34 (s, 2H), 3.86 (s, 3H), 3.81 (s, 3H), 13C NMR (75 MHz, CDCl3) δ: 161.3, 159.4, 156.1, 142.6, 135.4, 130.7, 127.5, 127.1, 126.6, 121.6, 120.9, 114.7, 114.4, 111.7, 55.4, 55.3, 48.2, Anal. calculated for C22H18Cl2N2O2(%): C: 63.87; H: 4.51; N: 6.87; Found: C: 63.91; H: 4.37; N: 6.72.
Conclusion
A new highly efficient methodology has been developed in THF solvent to generate N-arylmethylbenzimidazoles. Different diamines were used, excellent results were found in each case. Operational simplicity, clean reaction procedure, easy purification has increased the brightness of this methodology and above all no side products were found at 90 oC temperature.
Acknowledgement
Authors are very much grateful to their Ph.D. supervisors for providing laboratory facility and for studying spectra.
Refrences
- Gamovskii, A. F.; Simonov, A. M. Russ. Chem. Rev., 1996, 35, 122.
CrossRef - Spasov, A. A.; Yozhitsa, I. N.; Bugaeva, L. I.; Anisimova, V. A. Pharm. Chem. J., 1999, 33, 232-243.
CrossRef - Roth, T.; Morningstar, M. L.; Boyer, P. L.; Hughes, S. H.; Buckheit, R. W.; Michejda, C. J. J. Med. Chem., 1997, 40, 4199-4207.
CrossRef - Fonseca, T.; Gigante, B.; Gilchrist, T. L. Tetrahedron., 2001, 57, 1793-1799.
CrossRef - Denny, W. A.; Rewcastle, G. W.; Baguley, B. C. J. Med. Chem., 1990, 33, 814-819.
CrossRef - Migawa, M. T.; Girardet, J. L.; Walker, J. A. J. Med. Chem., 1998, 41, 1242-1251.
CrossRef - Valdez-Padilla, D.; Rodríguez-Morales, S.; Hernández-Campos, A.; Hernández-Luis, F.; Yépez-Mulia, L.; Tapia-Contreras, A.; Castillo, R. Bioorg. Med. Chem., 2009, 17, 1724-1730.
CrossRef - Sondhi, S. M.; Singh, N.; Kumar, A.; Lozach, O.; Meijer, L. Bioorg. Med. Chem., 2006, 14, 3758-3765.
CrossRef - Evans, B. E.; Rittle, K. E.; Bock, M. G.; Dipardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundel, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S.; Chang, R. S. L.; Lotti, V. J.; Gerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem., 1988, 31, 2235-2246.
CrossRef - Perston, P. N. Chem. Heterocycl. Compd., 1981, 40, 1-285.
- (a) Huang, W.; Scarborough, R. M. Tetrahedron Lett., 1999, 40, 2665.
CrossRef - (b) Dudd, L. M.; Venardou, E.; Garcia-Verdugo, E.; Licence, P.; Blake, A. J.; Wilson, C.; Poliakoff, M. Green Chem. 2003, 5, 187.
CrossRef - (a) Wang, Y.; Sarris, K.; Sauer, D. R.; Djuric, S. W. Tetrahedron Lett., 2006, 47, 4823-4826
CrossRef - (b) Lin, S. Y.; Isome, Y.; Stewart, E.; Liu, J. F.; Yohannes, D.; Yu, L. Tetrahedron Lett., 2006, 47, 2883-2886.
CrossRef - P¨atzold, F.; Zeuner, F.; Heyer, T.; Niclas, H.J. Synth Commun., 1992, 22, 281-288.
CrossRef - Stephens, F. F.; Bower, J. D. J. Chem. Sci., 1949, 71, 2971-2972.
CrossRef - (a) Mayer, J. P.; Lewis, G. S.; McGee, C.; Bankaitis-Davis, D. Tetrahedron Lett., 1998, 39, 6655-6658.
CrossRef - (b) Koprowska-Ratajska, M.; Kluczyk, A.; Stefanowicz, P.; Bartosz-Bechowski, H.; Szewczuk, Z. Amino Acids., 2009, 36, 309-315.
CrossRef - (a) Carpenter, R. D.; Lam, K. S.; Kurth, M. J. J. Org. Chem., 2007, 72, 284-287.
CrossRef - (b) Mao Z. Z.; Wang, Z. Y.; Li, J. N.; Song, X. M.; Luo, Y. F. Synth Commun., 2010, 40, 1963-1977.
CrossRef - Dabiri, M.; Salehi, P.; Baghbanzadeh, M.; Shakouri, N. M. Synth Commun., 2008, 38, 4272-4281.
CrossRef - (a) Perry, R. J.; Wilson, B. D. J. Org. Chem., 1993, 58, 7016-7021.
CrossRef - (b) Perumal, S.; Mariappan, S.; Selvaraj, S. Arkivoc., 2004, 8, 46-51.
(c) Salehi, P.; Dabiri, M.; Zolfigol, M. A.; Otokesh, S.; Baghbanzadeh, M. Tetrahedron Lett., 2006, 47, 2557-2560.
CrossRef - Singh, M. P.; Sasmal, S.; Lu, W.; Chatterjee, M. N. J. Heterocycl. Chem., 2014, 10, 1380.
- Maiti, D. K.; Halder, S.; Pandit, P.; Chatterjee, N.; Joarder, D. D.; Pramanik, N.; Saima,Y.; Patra, A.; Maiti, P. K. J. Org. Chem., 2009, 74, 8086-8097.
CrossRef - Naeimi, H.; Heidarnezhad, A. J. Chin. Chem. Soc., 2014, 61, 1004-1008
CrossRef - Luu, H.T.; streuff, J. Eur. J. Org. Chem., 2019, 1, 139-149
CrossRef - Huetling A.; Doye S. J. Org. Chem., 2002, 67, 1961-1964
CrossRef - Varala, R.; Nasreen, A.; Enugala, R.; Adapa, S. R. Tetrahedron Lett., 2007, 48, 69.
CrossRef - Sun, P.; Hu, Z. J. Het. Chem., 2006, 43, 773.
CrossRef - Mukhopadhyay, C.; Datta, A.; Butcher, R. J.; Paul, B. K.; Guchhait, N.; Singha,
- R. Arkivoc., 2009, (xiii), 1-22.
CrossRef - Salehi, P.; Dabiri, M.; Zolfigol, M. A.; Otokesh, S.; Baghbanzadeh, M. Tetrahedron Lett., 2006, 47, 2557.
CrossRef - Chakraborty, M.; Karmakar, S.; Mukherjee, A.; Arima, S.; Harigaya, Y. Heterocycl., 2006, 68, 967.
CrossRef - Kumar, B.; Kumari, S.; Kumar, B.; Cumbal, L. J. Chem. Sci., 2014, 126, 1831-1840.
CrossRef - Mohammadizadeh, M. R.; Taghavi, S. Z. Eur. J. Org. Chem., 2011, 8, 101-106.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


