A Review Article on Pharmaceutical Analysis of Pharmaceutical Industry According to Pharmacopoeias
Sony Ahmed1, Md. Shafiul Islam2*, Borkat Ullah2,3, Saurav Kanti Biswas1, Md. Abdus Samad Azad2 and Md. Shahadat Hossain2
1Department of Quality Control, Beximco Pharmaceuticals Ltd. (US FDA Approved Pharmaceutical Company), Gazipur-1700, Bangladesh.
2Department of Applied Chemistry & Chemical Engineering, Noakhali Science and Technology University, Noakhali, Bangladesh-3814
3Department of Chemistry, Virginia Commonwealth University, Richmond, VA, USA-23284
Corresponding Author E-mail: shafiulacce@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/360101
Article Received on : 12 Jan 2020
Article Accepted on : 18 Feb 2020
Article Published : 18 Feb 2020
The pharmaceutical tablet must meet specific standards to claim it as a standard drug approval. Pharmaceutical industries test the tablets for maintaining their accuracy following different standard parameters such as identity, strength, quality, purity, and stability, etc. For what why, it is essential to control pharmaceutical processes regardless of the issues that may be addressed. Process control includes inspecting raw materials, controlling processes, and targeting for the finished product. That’s why it is significant to monitor the effectiveness of the process control. In connection to this, the adaptation of the production process should comply with the specification as needed, which may also include control of equipment and environment. Pharmaceutical products in the process should be checked appropriately for their identity, strength, quality, and purity and the products are approved or rejected by the quality control unit during the manufacturing process. The highlights of this review are to describe quality control testing of pharmaceutical products by using different instruments for the pharmaceutical industry, according to pharmacopeias.
KEYWORDS:Finished Product; Purity; Pharmacopeias; Raw Material; Quality Control
Download this article as:| Copy the following to cite this article: Ahmed S, Islam Md. S , Ullah B, Biswas S.K , Azad Md. A. S, Hossain Md. S. A Review Article on Pharmaceutical Analysis of Pharmaceutical Industry According to Pharmacopoeias . Orient J Chem 2020;36(1). |
| Copy the following to cite this URL: Ahmed S, Islam Md. S , Ullah B, Biswas S.K , Azad Md. A. S, Hossain Md. S. A Review Article on Pharmaceutical Analysis of Pharmaceutical Industry According to Pharmacopoeias . Orient J Chem 2020;36(1). Available from: https://bit.ly/2vGEHgA |
Introduction
The pharmaceutical industry is one of the most regulated industries worldwide because the drugs produced must be safe and effective. The Food and Drug Administration (FDA) requires that raw materials are tested before manufacturing pharmaceutical products to establish their identity, purity, and quality. This analysis is an essential step in the production of pharmaceuticals and ensures that the product is suitable for its intended use. Our capabilities include testing raw materials, Active Pharmaceutical Ingredients (APIs), excipients, and various additive based finished products, including United States Pharmacopeia (USP) /National Formulary (NF), Japanese Pharmacopoeia(JP), European Pharmacopoeia(EP), Food Chemical Codex(FCC) and British Pharmacopoeia(BP). Quality is not an accident; it is the result of sensible efforts1.Synthesis and the properties and properties of such molecules, known as active pharmaceutical ingredients (APIs), and their analysis and therapeutic efficacy data are prerequisites for drug candidate identification2. The regulatory authority permits clinical trials after passing pre-clinical tests for clinical trial on the proposed system drug. The clinical trial test produces statistically significant data on the association of the drug’s risky performance, safety, and overall benefit. At the same time, the drug is working on the proposed system, its optimal dose, and schedule. This step determines the drug’s potential interaction with other drugs and monitors the long-term viability of the drug. After successful completion of the clinical trial, the drug comes to the market for patients. Various guidelines related to chiral drugs have been published that encourage the development of a single enantiomer drug for pharmaceutical manufacturers3,4. Registration for quality pharmaceuticals for chiral drugs was determined by the International Conference on the Harmonization of Technical Requirements in the Human Use for Pharmaceuticals Registration5. Quality has become a critical and sensitive issue in the pharmaceutical industry. As the World Food and Drug Administration (FDA) for the twenty-first century has come together to introduce, practice and guide and integrate the current Good Manufacturing System (cGMP), there is an increasing awareness of the importance of pharmaceutical products. Pharmaceutical finished products may contain particles of unknown foreign substances6. Foreign issues should be identified, and its source should be defined to prevent further contamination. In the pharmaceutical industry, it is essential to control the defects at every stage of the manufacturing process. The total quality of the product must be ensured according to the combination of the drugs7. Manufacturing practices that result in the production of good quality finished products and adequate considerations to protect employees are recognized as Good Manufacturing Practice (GMP). GMP is associated with both production and quality control (QC)8. QC is the part of GMP whereby QC staff analyzes the quality of all factors involved in production to remove defects at each stage of production. The purpose of QC is to produce a perfect finished product by preventing defects at each stage of production. QC is teamwork, and we must remember that quality must be created as a drug product when designing a product and process. It is impacted by physical plant design, placement, ventilation, cleanliness, and sanitation during routine production9. In process quality control testing is performed at regular time intervals during the manufacturing process10. The objectives of the IPQC include monitoring and modifying the production process to ensure compliance with the requirements. The process control can involve the environment as well as equipment control. They should not take any risk of product quality. The process helps identifying the problems in the test easily. It sometimes identifies a defective product batch that can be repaired, but once this batch is finished contrariwise it may not be possible. Failure to meet the IPC specification indicates that either those procedures were not followed or that some factors were out of control11. Standard operating procedures (SOPs) should be established in the pharmaceutical industry, and then IPQCs and tests are described12. The highlight of this review is to show the quality parameters of pharmaceutical analysis according to pharmacopeias, which are part of the raw materials and finished product for quality control tests.
Pharmaceutical Analysis for Quality Control Testing in Pharmaceutical Industry
The evaluation of pharmaceutical raw materials and finished products for impurities and degradation products is an essential part of the process of drug development and production. Furthermore, toxicity data must be found on any drug-related impurities that represent a concentration of more than 0.1% of the active pharmaceutical ingredient (API). Conventional analysis of pharmaceutical QC and production fields has traditionally been performed by titration, Identification, Loss on drying, sulfate ash, dissolution, and disintegration test by using UV-visible, HPLC, GC, or IR detection.
Universal Tests for Pharmaceutical Industry
Some tests that are generally applicable to the pharmaceutical tablet and other pharmaceutical products.
Description
According to Loyd V Allen Jr. (2013), conduction of these tests likely to present the specification and is a qualitative description of a pharmaceutical tablet. For example, a specification’s tablet description reads white, off-white, round, biconvex, film-coated tablet, painted with “Rx” on one side13.
Identification
The purpose of identification testing is to verify the identity of the active pharmaceutical ingredient (API) on the pharmaceutical tablet. The identification test will be able to discriminate between compounds of nearly related structures that are probably present. Identification tests should be specific to new drug substances, for example, infrared spectrum.
Assay
A specific, stability-indicating assay test to determine the strength (content) of the API in pharmaceutical tablets. In many cases, it needs to apply the same method (for example, UV / HPLC are shown in figure 1) for both the drug substance and the number of impurities.
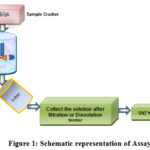 |
Figure 1: Schematic representation of Assay |
Chromatography and spectroscopy are orthogonal techniques meaning that both types of technique provide different and specific information. Chromatography is a kind of separation method and spectroscopy is a technique that gives a ‘fingerprint’ to individual or molecular mixtures. HPLC is a technique that assist for separation, quantification, and element detection in mixtures. It is especially suitable for compounds that are not easily volatile, are not thermally unstable, and have high molecular weight. The merit of UV method over HPLC method is that UV method does not usually require elaborate treatments and procedures related to chromatographic method. It is time consuming and economically low. The HPLC and UV spectrometry method is an adequate method of measuring a drug in pure form and its dose. Because these methods are simple, fast, precise and accurate, they can be successfully and conveniently adopted for quality control analysis of drugs in the form of bulk and pharmaceutical doses on a routine basis.
Assay and Content Uniformity
Drug Impurities Analysis
Drug discovery & Drug development
Method Development and Validation of Drugs
UV Absorption Spectroscopy Analysis for Pharmaceutical Industry
Detection of Impurities
UV absorption spectroscopy for determining impurities in organic molecules is one of the best methods. Extra peaks can be monitored for impurities remain in the sample and can be compared with standard raw materials. Impurities can also be detected by measuring absorption at specific wavelengths. For example, Benzene appears as a common impurity in cyclohexane. Its presence is easily detectable by absorbance of 255 nm.
Quantitative Analysis
The UV absorption spectrum can be used to quantitatively determine the compounds that absorb UV radiation. This determination is based on the law of beer as below
A = log I0 / It = log 1/ T = – log T = abc = εbc
Where ε is extinction co-efficient, c concentration, and b is the length of the cell used on the UV spectrophotometer. UV-3000 UV / Vis spectrophotometer with a 1 cm match quartz cell to holographic grating system, observed light of the instrument is reduced and the analysis occur more accurate. Pharmaceutical product is also known for its stable performance by using UV / Vis spectrophotometer.
Qualitative Analysis
UV absorption spectroscopy can identify the types of compounds that absorb UV radiation. The absorption spectrum is identified by comparing the spectrum of the known compound. The UV absorption spectrum is commonly used to identify aromatic compounds and aromatic olefins
Quantitative Analysis of Pharmaceutical Substances
Drugs can be either in the form of raw materials or in formulas. These can be synthesized by creating appropriate drug solutions in the solvent and measuring absorption at specific wavelengths is shown in figure 2. The Diazepam tablet can be analyzed by methanol by 0.5% H2SO4 at wavelength 257 nm. Qualitative analysis through spectrophotometric methods yields fast and accurate results using only small sample volumes. This fast and efficient material has become an indispensable tool in the pharmaceutical industry, thanks to its adaptability and economic value. Qualitative analysis has proven to be highly effective at many large levels of organic compounds and this helps to ensure the health and safety of the patient.
 |
Figure 2: UV Absorption spectroscopy Analysis |
High-Performance Liquid Chromatography Analysis for Pharmaceutical Industry
High-performance liquid chromatography (HPLC) is a chromatographic analytical technique used to split a mixture of chemistry, biochemistry and industrial field compounds. The main purpose for using HPLC is to purify the quantities and mix individual components. Since it is used to test products and identify the raw material, it is also used to perform qualitative and quantitative analysis of their products. HPLC plays a significant and critical role in the pharmaceutical industry and analysis. An HPLC instrument includes a pump, injector, column, and detector and information acquisition and display system. It can detect small amounts of solvent & improved resolution of the compound from the column. Furthermore, the importance of HPLC in this field is subject to stringent regulations established by the Food and Drug Administration (FDA) in the United States. According to the USA Pharmacopoeia (1999), 14the HPLC method established for the first time the trial of bulk drug materials. It makes analytical support within the pharmaceutical industry15 become the preferred method of quality control and assurance at many levels. In addition to HPLC analysis of the drugs and its application to impurities analysis of the pharmaceuticals has been applied16,17.All these pharmaceutical companies apply this compulsory method HPLC to identify the quality of their products before allowing them to be sold in international markets. The most important benefit of using HPLC technique in the industry and analytical field is that it helps to explain the structure and quantitative determination of impurities and degradation products in large quantities of drug materials and pharmaceutical composition. This benefits from the use of HPLC is limited not only to synthetic drugs and formulas but also to include herbal medicines too. Drug discovery, development, and production utilize widely used high performance liquid chromatography (HPLC) to separate individual compounds for the detection, quantification, and purification of different components. Schematic Representation of HPLC analysis are shown in figure 3
 |
Figure 3: High-performance liquid chromatography Analysis |
Thus, there is a great demand for HPLC instruments for advanced analysis of the “next generation” detection. Monica Young and Mark at Eksigent Corporation have tested the effectiveness of Eksigent Express LC Ultra Micro-High-Pressure Liquid Chromatography for a real-world pharmaceutical application. By combining the capability of micro-flow HPLC, and the function to separate under high pressures, the new instrument enables fast separation efficiency, cost reduction and high system reproducibility18. Most workers use reverse-phase mode with UV absorption detection when appropriate, because it provides the best available reliability, analysis time, repeatability and sensitivity. HPLC has been used in several pharmaceutical drug manufacturers 19-22 and biological fluids 23-26. As such, HPLC provides an excellent service in answering many of the questions raised by the pharmaceutical industry. However, the limitations of HPLC lack the long-term reproducibility due to the proprietary nature of column pricing, solvent and column packing. Liquid chromatography combined with mass spectrometry (LC-MS) is considered to be one of the most important analytical technique of the last decade of the 20th century 27. The pharmaceutical industry has become the preferred method for quality control and analytical support at many levels of assurance 28-29. HPLC-MS has recently been used for drugs 30-36. HPLC drugs have been applied for the analysis of drugs alone, as well as for the pharmaceuticals 37-41 and the impurity products 42 – 44.
Impurities
Organic as well as inorganic impurities and residual solvents are included in this section. This test determines the presence of any component that is not API or an excipient to pharmaceutical products. The most common types of impurities that are measured are related substances, which are impure processing of new drug substances, API degradation products.
Raw Materials and Finished Product Test for Pharmaceutical Industry
The physical parameters test of raw material for the pharmaceutical industry are temperature, pressure, moisture content, weight, particle size, hardness, loss of drying, sulfate ash, color, integrity. Test of the finished product for pharmaceutical industry are assay, Uniformity of content, uniformity of mass, weight variation, friability test, content of active ingredients, hardness test, disintegration test, dissolution test etc. The raw materials and finished product tests for the pharmaceutical industry according to pharmacopoeias are listed below
Size and Shape
The size and shape of the tablet can be described dimension-ally and controlled. This is determined by the equipment during the condensation process45.
Color and Odor
Many pharmaceutical tablets use color as an essential means of quick identification and consumer acceptance. But it certainly equates to a lot in a single tablet, from tablet to tablet and lot. The presence of odor in a batch of tablets may indicate stability issues such as. Vitamins may have a characteristic odor, for example, the characteristic odor of acetic acid or the drug’s ability to degrade aspirin tablets. Flavor tablets are important in customer acceptance45.
Moisture Content of Granules
The granules should have enough strength to not break a large amount of fine powder and prevent normal handling and mixing processes. On the other hand, it is wise to expose some areas of clear surfaces for optimal bonding while reducing the size of the connections on the tablets, so moisture content is very important for producing good drug products45.
Assay
A tablet contains an active ingredient called API. So in order to be able to make the tablet well finished product relations production has to be done using proper analysis method46.
Water content:
Acceptance criteria may be justified with information on the effects of hydration or water absorption on drug products. In some cases, a loss may be considered sufficient in the drying process; however, there is an identification method that is specific for water (e.g., Karl Fisher titration) are shown in figure 4
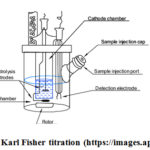 |
Figure 4: Karl Fisher titration |
Many active pharmaceutical ingredients (APIs) and adjuvants are water or bound in an adsorbed form (surface water) called hydrate. The water content of drugs strongly affects their quality, shelf life and durability as well as the release of active substances. Water determination is therefore an important assumption in pharmaceutical analysis. By far, the most important method for determining the water content of drugs is Karl Fisher titration. The Karl Fisher oven method is suitable for samples that simply release their water at high temperatures, difficult to dissolve or react with KF reagent. Sample preparation is very easy. The material to be analyzed is loaded into a sample vial, which is hermetically sealed with a septum and placed on a rack. Heat the sample in an oven and release water from the sample. A double blank needle pores the septum and a stream of carrier gas transports moisture to the titration cell for subsequent determination.
Uniformity of Content
A physically protected tablet may not produce the desired effect. To evaluate tablet potential for efficacy, it is necessary to monitor the amount of drug per tablet, from tablet to tablet, and from batch to batch. For this test according to BP using the appropriate experimental analysis method, determine the individual content of active substance of 10 tablets taken at random46. The tablet complies with BP-based testing, if the average content of each individual content is 85 percent to 115 percent. If the tablet fails to comply with the test, more than one distinct content is out of range or if one individual content is out of the range the average content is 125 percent to 75 percent. If an individual content exceeds the 85 percent to 115 percent limit, set the individual content of the other 20 tablets taken randomly, in the range of 75 percent to 125 percent. The tablet agrees with the test if no more than 30 tablet-specific content is out of the range of 85 percent to 115 percent of the average content and nothing is out of the range of 75 percent to 125 percent of the average content46.
Microbial limit
Microbial limit is seen as a feature of experimental good manufacturing Practice, as well as quality assurance. In general, it is advisable to test the product of the drug unless its ingredients are tested before production and the manufacturing process is informed, through validation studies, not carrying a significant risk of bacterial infection or outbreak. It should be noted that this guideline does not directly address externalities, but the policies outlined here may apply to outsiders as well. In both cases, skip testing can be an appropriate method. With acceptable scientific justification, it may be possible to suggest a microbial limit test for powders intended for reconstitution as oral liquids.
Uniformity of dosage units
This term includes both the mass of the dosage form and the content of the active substance in the dosage form; a pharmacopoeia approach should be used. In general, specifications should include one or the other, but not both. If appropriate, this can be edited in the testing process. Acceptance criteria should be included in the specification. When applying weight variations to new drug products in pricing, applicants should verify that the homogeneity of the product is sufficiently sufficient when developing the drug
Weight Variation Test
According to the USP Weight variation test, up to 20 tablets are independently run by comparing average weight and average individual tablet weight. Weight loss tests are manifested in percentages. The following formulas are used:
Weight Variation = (Iw –Aw)/Aw ´ 100% where, Iw = Individual weight of tablet; Aw = Average weight of tablet47.
According to the USP, the tablet agrees to the test if the individual population has no more than 2 percent deviations from the average mass, as shown in Table 1, and no more than twice the percentage47.
Table 1: USP limits for weight variation test for uncoated tablets
| Average Weight (mg) | Percentage Deviation (%) |
| 130 or Less | 10 |
| 130 – 324 | 7.5 |
| More than 324 | 7 |
Hardness Test
One of the early testers for this test was the Ketan Tablet Hardness Tester, a type of Monsanto hardness tester for evaluating the tablet hardness tester. The tester has a barrel. Lower plunger is kept in touch with tablet and zero read is taken. The threaded bolt is forced against a spring until the upper plunger enthusiast cracks the tablet. As the spring narrows, a pointer moves with the barrel gauge to indicate the ball. The force of the crack is recorded in kilograms. It is naturally appropriate to examine the rigidity as a procedural control. In these situations, it is usually not necessary to include these features in the specification. If the hardness characteristics have a serious impact on the quality of the product of the drug (e.g., chewable tablets), the standard of recognition should be included in the specification47.
Disintegration Test
The USP disintegration apparatus consists of 6 glass tubes that are 3 inches long, open at the top, and placed at the bottom end of the basket rack assembly against the 10-mesh screen, with one tablet placed on each tube, and the basket rack fixed at medium 37± 2 ° C such tablets are located 2.5 cm below the surface of the fluid at their upward motion and the vessel descends closer than 2.5 cm from the bottom. A standard motor-driven device is used to move the basket assembly with tablets at distances of 5 to 6 cm at a frequency of 28 to 32 cycles per minute. Perforated plastic disks can also be used in the tests. These are placed on top of the tablets and provide a harmful action on the tablets. While disks can be meaningful or create more sensitivity to tests, they are useful for floating tablets. Operate the device for a fixed time (15 minutes for uncoated tablets unless otherwise justified and approved) 47. The tablet complies with the test, if the tablets are disconnected and all particles pass through the 10-mesh screen at the specified time. If any residue is left, it must have a soft mass that has no clear firm core. The tablet complies with the experiment according to USP, if all tablets are completely disconnected. If 1 or 2 tablets fail to break completely, repeat the test on 12 additional tablets. Total 18 tablets not less than 16 but the requirements are met to test The British Pharmacopoeia (BP) and Indian Pharmacopoeia (IP) limits for disintegration times of tablets are given in Table 2, Table 3 and shown in figure 5 respectively47.
Table 2: BP limits for disintegration times of tablets
| Categories of Tablets | Disintegration Time (min) |
| Uncoated tablets | 15 |
| Coated tablets | 60 |
| Effervescent tablets | 5 |
| Soluble tablets | 3 |
| Dispersible tablets | 3 |
| Oro dispersible tablets | 3 |
| Gastro-resistant tablets | 60 |
| Oral lyophilisates | 3 |
Table 3: IP limits for disintegration times of tablets
| Categories of Tablets | Disintegration Time (min) |
| Uncoated tablets | 15 |
| Coated tablets | 60 |
| Enteric-coated tablets | 60 |
| Film-coated tablets | 30 |
| Effervescent tablets | 5 |
| Soluble tablets | 3 |
| Dispersible tablets | 3 |
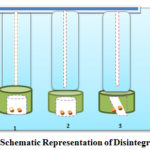 |
Figure 5: Schematic Representation of Disintegration Tester |
Dissolution Test
BP or USP dissolution apparatus (paddle / basket apparatus) consist of a cylindrical vessel with a hemispherical bottom, which may be covered with glass or other inert, transparent material; a motor; a metal drive alloy; and a cylindrical basket are shown in figure 6. The vessel is partially immersed in a convenient water bath of a convenient size or heated by a suitable device such as a heating jacket. A water bath or heating device allows the temperature inside the vessel to be kept at 37 ± 0.5 ° C during the test and keeps the bath fluid in constant, smooth motion. According to BP, keep the specified volume (± 1 %) of the dissolution medium in the vessel for this test46. Assemble the apparatus, adjust the dissolution medium to 37 ± 0.5 ° C. Carefully remove 1 of the air bubbles from the surface of the tablet. Operate the apparatus at a fixed rate. Within a specified time interval, or each time specified, withdraw a sample from the zone of the midway between the surface of the dissolution medium and the top of the rotating basket, not less than 1 cm from the wall of the vessel. Where multiple sampling times are specified, replace the withdrawn aliquots at 37 ° C for newly dissolution medium or, where it can be shown that the medium does not need replacement, is correct for volume changes in the calculation. Keep the vessel covered for the duration of the test, and at a suitable time, check the temperature of the medium. Perform analysis using appropriate side procedures as directed by individual monographs. Repeat the experiment with additional tablets. Unless specified otherwise in separate monographs, 5the amounts of active ingredient dissolved in the tablets tested from the tests required by British Pharmacopoeia(BP), United States Pharmacopeia (USP), European Pharmacopoeia (PhEur), Japanese Pharmacopoeia(JP) and International Pharmacopoeia (PhInt) meet the following recognition criteria are show in table 4.
Table 4: BP, USP, PhEur, JP and PhInt acceptance criteria for dissolution test of tablet
| Stage | Number of Tablet Tested | Acceptance Criteria |
| S1 | 6 | Each unit is not less than Q + 5 %. |
| S2 | 6 | Average of 12 units (S1 + S2) is equal to or greater than Q, and no unit is less than Q – 15 %. |
| S3 | 12 | Average of 24 units (S1 + S2 + S3) is equal to or greater than Q, not more than 2 units are less than Q – 15 %, and no unit is less than Q – 25 %. |
If the results are not consistent with S1 or S2, continue the experiment in phase 3 Quantity Q, is the specific amount of dissolved active substance, expressed as a percentage of the labeled material; the 5 percent, 15 percent, and 25 percent values in the table are the percentage of content labeled so that these values and Q are in the same terms 46.
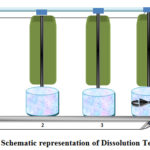 |
Figure 6: Schematic representation of Dissolution Tester |
Conclusion
The main goal of pharmaceutical products is to serve human beings to protect them from potential illness or disease. The drug should be free from impurities or other interference to fulfill its intended purpose, which can harm people. Pharmacopeias sets the standard for high quality drugs. According to the WHO list, 140 independent countries currently employ around 30 national and African, European and international pharmacopoeias. From present study, it is clearly expressed that various pharmacopoeias suggest different types of raw materials and finished products test for pharmaceutical tablet using different instruments in the pharmaceutical industry, but the main purpose of pharmacopoeias worldwide is to produce good quality drugs for human health.
Acknowledgements
The authors would like to thank, Deputy general manager, Department of quality control (Track-1), Beximco pharmaceutical (US FDA Approved Pharmaceutical company), Gazipur-1700, Bangladesh for giving of necessary information, helpful comments and other facilities for this review.
Conflict of Interest
The author declares no conflict of interest
Author’s Contributions
This is done with the cooperation of all the authors. The authors designed the review study, wrote the protocol, reviewed the scientific content of the manuscript, conducted the analysis of the study, and drafted the manuscript. All authors read and approved the final manuscript.
References
- Dewan, S.M.R.; Alam, A.; Ahamed, S.K. Int. Res. J. of Pharm, 2013, 4, 96.
- Valagaleti, R.; Burns, P.K.; Michael, G. Analytical Support for Drug Manufacturing in the United States—From Active Pharmaceutical Ingredient Synthesis to Drug Product Shelf Life. Drug Information Journal, 2003, 37, 407-438
CrossRef - FDA’s Policy statement for the development of new stereoisomeric drugs, 1992, 4, 338–340
CrossRef - Health Canada, 2000. https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/hpfb-dgpsa/ pdf/ prodpharma/ stereo-eng.pdf
- European Pharmacopoeia Commission. European Pharmacopoeia, 8th Edition, Council of Europe, Europe, 2013.
- Sony, A.; Anwar, M.H.; Shafiul, I.; Islam, M.M. A review article on introduction of analytical instruments analysis in pharmaceutical industry according to pharmacopoeia, Mintage Journal of Pharmaceutical and Medical Sciences.2019, 1-4.
- Woodcock, J. Ameri. Pharmace. Rev. 2004, 7, 10-15.
- Fortunak, J.M.; Souza, R.D.; Kulkarni, A.A.; King, C.L.; Ellison, T.; Miranda, L.S.M. Antivi. Thera, 2014, 9, 4-6.
- Mehmood, T.; Salaria, M.R.; Herani, G.M.; Qureshi, M.A. Ind. J. of Manage. & Soci. Sci. 2009, 3, 21-30.
- Levi, L; walker, G. Canadi. Medi. Associ. 2010, 91, 96.
- Tangri, P; Mamgain, P; Shaffi; Verma, A.M.L; Lakshmayya. Int. J. of Ind. Pharm. and Bio Sci, 2012, 1, 49-51.
- Mazumder, B; Bhattacharya, S; Yadav, A. Int. J. of Pharm Tech Res. 2011, 3, 366.
- 13. Loyd,V.A.J. Remington Introduction to Pharmacy, 1st Edition, Pharmaceutical Press, UK. 2013, 146
- The United States pharmacopeia; the national formulary United States Pharmacopeial Convention. USP Convention Inc., Rockville, 1999
- White, D.; Varlashkin, P.; Rusch, D.N. ‘A thin-layer chromatographic method to determine process impurities in leucovorin calcium. J Pharm Sci. 1992, 81, 1204-9.
CrossRef - Nicolas, E.C.; Scholz, T.H. ‘Active drug substance impurity profiling part II. LC/MS/MS fingerprinting. J Pharm Biomed Anal. 1998, 16, 825-36
CrossRef - Madireddy, V.; Babu, K.S.; Narayanreddy, P.G. J. Anal. Chem., 2011, 2, 198–207
- Monica, M. Y.; Xiaoyi, G.; Schafer, W.; Arnold, D.; Christopher, J. Welch Evaluation of micro ultra-high pressure liquid chromatography for pharmaceutical analysis. Anal. Methods. 2013, 5, 2178-2181
CrossRef - Siddiqui, M.R.; Tariq, A.; Reddy, K.D.; Chaudhary, M.; Yadav, J.; Negi, P.S.; Bhatnagar, A.; Singh, R.. Int. J. Pharmacol. 2010, 6, 271–277
CrossRef - Tang, J.; Peng, J.; Zhang, L.; Xiao, X. Anal. Methods. 2012, 4, 1833– 1837
CrossRef - Devika, G.S.; Sudhakar, M.; Rao. J.V., 2012, 9, 999– 1006.
CrossRef - Ahmed, M; Manohara, Y.N; Ravi, M.C., Int. J. Chem. Technol. Res. 2012, 4, 337–345
- Tariq, A.; Siddiqui, M.R.; Kumar, J.; Reddy, D.; Negi, P.S.; Chaudhary, M.; Srivastava, S.M.; Singh, R.K. Sci. Asia , 2010, 36, 297–304.
CrossRef - Samanidou, V.; Pantazidou, K.; Kovatsi, L.; Njau, S.; Livanos, A. J. Sep. Sci. 2012, 35, 839–845.
CrossRef - Malenovic, A.; Jovanovic, M.; Petrovic, S.; Kostic, N.; Vemic, A.; Janc. S.B. Inst. Sci. Technol. 2012, 40, 138–149.
CrossRef - Giorgi, M.G.; Howland, K.; Martin, C.; Bonner, A.B. A novel HPLC method for the concurrent analysis and quantitation of seven water-soluble vitamins in biological fluids (plasma and urine. Sci. World J. 2012,
CrossRef - Niessen, W.M.A. J. Chromatogr. 1999, 179–197.
CrossRef - Ermer, J., J. Pharm. Biomed. Anal. 1998, 18, 707–714.
CrossRef - Nicolas, E.C.; Scholz, T.H. J. Pharm. Biomed. Anal. 1998, 16, 825– 836.
CrossRef - Hilhorst, M.J.; Hendriks, G.; van Hout, M.W.; Sille´n, H.; van de Merbel, N.C. Bio-analysis 2011, 3, 1603–1611.
CrossRef - D’Avolio, A.; Simiele, M.; Siccardi, M.; Baietto, L.; Sciandra, M.; Bonora, S.; Di Perri, G. J. Pharm. Biomed. Anal. 2010, 52, 774– 780.
CrossRef - Berset, J.D.; Brenneisen, R.; Mathieu, C. Chemosphere. 2010, 81, 859–866
CrossRef - Ding, L.; Huang, X.; Yang, J.; Bian, X.; Zhang, Z.; Liu, G. J. Pharm. Biomed. 2006, 40, 758-62
CrossRef - Wren, S.A.C.; Tchelitcheff, P. J. Pharm. Biomed. Anal. 2006. 40, 758–762.
CrossRef - Zhou, J.; Dai, X.; Fang, X.; Li, H.; Zhao, Y.; Gong, A. Instrum. Sci. Technol. 2011, 39, 522–533
CrossRef - Breaud, A.R.; Harlan, R.; Kozak, M.; Clarke, W. Clin. Biochem. 2009, 42, 1300–1307.
CrossRef - Chitturi, S.R.; Somannavar, Y.S.; Peruri, B.G.; Nallapati, S.; Sharma, H.K.; Budidet, S.R.; Handa, V.K.; Vurimindi, H.B. J. Pharm. Biomed. Anal. 2011, 55, 31–47.
CrossRef - Madireddy, V.; Babu, K.S.; Narayanreddy, P. Glob. J. Anal. Chem. 2011, 2, 198–207.
- Navaneeswari, R.; Reddy, P.R. Afr. J. Scient. Res. 2011, 6, 318–324.
- Thomas, S.; Bharti, A.; Tharpa, K.; Agarwal, A. J. Pharm. Biomed. Anal. 2012, 60, 86–90.
CrossRef - Verbeken, M.; Suleman, S.; Baert, B.; Vangheluwe, E.; Dorpe, S.V.; Burvenich, C.; Duchateau, L.; Jansen, F.H.; De Spiegeleer, B. Malaria J. 2011, 10, 51. 51
CrossRef - Sabry, S.M.; Belal, T.S.; Barary, M.H.; Ibrahim, M.E.A. Drug Test Anal. 2012.
- Hanysova, L.; Grafnetterova, T.; Dubovska, B.; Klims, J. Chem. Papers 2005, 52, 99–102
- Bouchonnet, S.; Bourcier, S.; Souissi, Y.; Genty, C.; Sablier, M.; Roche, P.; Boireau, V.; Valerie, I. J. Mass Spectrum. 2012, 47, 439–452
- Lachman, L; HA Lieberman, H.A.; Kanig, JL. The Theory and Practice of Industrial Pharmacy, 3rd Edition. 1986; 296-300.
- 46 British Pharmacopoeia Commission; British Pharmacopoeia, 13th Edition. 2013.Unites States Pharmacopoeia Convention; United States Pharmacopoeia. 38-National Formulary 33, 2010.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


