Theoretical Study for the [2+2] Cycloaddition Reaction Mechanism of Ketenes and their Derivatives
Haydar A. Mohammad-Salim1and Hassan H Abdallah2
1Department of Chemistry, Faculty of Science, University of Zakho, Duhok 42001, Iraq
2Department of Chemistry, College of Education, Salahaddin University, Erbil 44001, Iraq
Corresponding Author E-mail: hwchems@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/350512
Download this article as:
![]()
This study presents the intramolecular [2+2] cycloaddition reaction of ketenes to form cyclobutanones using B3LYP-D3/6-311++G(d,p) level of approximation. The concerted mechanism path was studied in detail. The structures of all intermediates and transition states were located using same level of theory. The influences of the substituents (-H, -CH3, -NH2, -F, -OH and –CN) were also discussed. The analysis of stationary points and the energetic parameters indicates that the substituted ketene with –CN group has the highest activation energy; however, ketene with –NH2 group has the lowest one. Conversely, in-tramolecular [2+2] cycloaddition records the highest degree of asynchronicity with –NH2 group and lowest with –CN group. The calculated thermodynamic parameters at room temperature have been listed and analyzed. The global and local properties of reactants involved in the intramolecular [2+2] cycloaddition reactions and the Fukui functions for an electrophilicity and local electrophilicity were also elucidated for carbon centers of each reactant.
KEYWORDS:DFT; Mechanism; Ketenes; [2+2] Cycloaddition
Introduction
One of the earliest ketenes applications in the synthesis of natural products employed the [2+2] cycloaddition reactions. 1 Cyclobutanones are considered a powerful synthetic intermediates involved in processes such as vicinal and geminal alkylation. 2 They are readily prepared by the reaction of olefins with keteniminium salts or activated ketenes. 3-4 The intramolecular form of these cycloadditions could offer promising paths for the stereo- and regio- controlled synthesis of polycyclic compounds. 5 The intramolecular stereospecific cycloaddition of ketenes to olefins is a significant method for the preparation of cyclobutanones 6 and this is one of the main methods for the carbofunctionalization of olefins.7 With ketenes itself, this cycloaddition proceeds in poor yield since ketenes are easily dimerized. Cycloaddition of ketenes containing electron withdrawing groups for instance sulfur, oxygen or halogens are more general.8 Recent studies obtained very good results with α,β-unsaturated ketenes.
The general form for an intramolecular [2+2] cycloaddition of ketene is presented in Scheme 1. The ketene is obtained from the conversion of a ketene precursor. Some of the most common methods used are zinc reduction of α-chloro acid chloride, Wolff rearrangement of α-diazo ketone and based-induced hydrogen chloride elimination from an acid chloride.9-11 Ring opening of cyclobutanones, 1,5-sigmatropic rearrangements of conjugated dienals, pyrolysis of esters, elimination from mixed anhydride and some special cases of photochemical fragmentation processes have been applied with excellent success.12-16
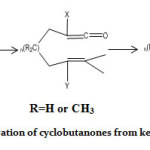 |
Scheme 1: Preparation of cyclobutanones from ketene precursor. Click here to view scheme |
The intramolecular [2+2] cycloaddition of ketene has been investigated experimentally by Marko and Ronsmans, 1985.17 Their studies involved cycloaddition of alkenylketenes R1 (see Scheme 2) derived from unsaturated acid chlorides. However, to the best of our knowledge, the detail information about the reaction mechanism and its stereo- and regioselectivity have not been obtained yet. Therefore, the aim of this paper is to suggest the concerted mechanism of the intramolecular [2+2] cycloaddition of alkenylketenes and its derivatives using DFT method. Herein, electron withdrawing and donating groups are used in order to examine the electronic effect of these substituents on the regioselectivity and the reaction mechanism.
![Scheme 2: Proposed Mechanism for the intramolecular [2+2] Cycloaddition reactions.](https://www.orientjchem.org/wp-content/uploads/2019/10/Vol35_No5_The_Hay_sch2-150x150.jpg) |
Scheme 2: Proposed Mechanism for the intramolecular [2+2] Cycloaddition reactions. Click here to view scheme |
Computational Methods
Gaussian 09 package under Linux operation system was used and all studied structures were fully optimized in the gas phase.18 DFT method has been proven to be a convenient method for the study of intramolecular [2+2] cycloaddition reactions and is employed in this study.19-21 The B3LYP functional is used throughout in combination with 6-311++G(d,p) basis set.22-23 We also considered the D3 correction to take into account for dispersion.24 Frequency calculations were performed to ensure that a transition state has only one imaginary frequency and a local minimum has no imaginary frequencies. Intrinsic reaction coordinate (IRC) computations were carried out to verify that the transition states connect the required reactants and products.25
All energies and thermodynamic parameters reported in this research were obtained from the frequency calculations at the same level of approximation. The Gibbs free energies, enthalpies and entropies in the gas phase were obtained with the standard statistical thermodynamics at 1 atm and 298.15 K. The CYLview software was used as a graphical interface.26
The global electrophilicity index (ω) is obtained on the basis of the electronic chemical potential (µ) and the chemical hardness (η) using the equation (1) 27:

The chemical hardness (η) and the electronic chemical potential (µ) quantities may be approached based on the one electron energies of Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO), εH and εL, as in equation (2) and (3) 28-29:

The relative nucleophilicity index (N) obtained in terms of the energies of HOMO (EHOMO(Nu)) within the scheme of Kohn-Sham.30 This quantity can be defined by using equation (4). Where TCE is tetracyanoethylene and is chosen due to its lowest HOMO energy as a reference.31
Condense Fukui functions and the related local parameters are significant to examine chemical reactivity in the center site of organic molecules.32 The Fukui functions are calculated using same level of theory. The Fukui functions for electrophilic attack, , and for nucleophilic attack, , allows the identification of the most nucleophilic and electrophilic centers in the organic species.
![Figure 1: Schematic structure and atom numbering of reactant involved in intramolecular [2+2] cycloaddition reaction of ketenes.](https://www.orientjchem.org/wp-content/uploads/2019/10/Vol35_No5_The_Hay_fig1.jpg) |
Figure 1: Schematic structure and atom numbering of reactant involved in intramolecular [2+2] cycloaddition reaction of ketenes. Click here to view figure |
Results and Discussion
The concerted mechanism of [2+2] intramolecular cycloaddtion of R1, R2, R3, R4, R5 and R6 is studied theoretically using B3LYP-D3/6-311++G(d,p) level of approximation. The transition state structures for the intramolecular cycloaddition were first located to examine the possibility of a concerted reaction mechanism. The IRC calculations illustrate the lowest energy pathway connecting the reactant to the cycloadducts (see Figure 2). This figure illustrates the IRC for the parent (unsubstituted) system. The C2-C3 bond distance was plotted versus the C1-C4 bond distance across the reaction pathway in a More O’Ferrall-Jencks type diagram (See Figure 3). The nature of asynchronous between the two forming bonds can be visualized via this diagram, which is clear from the deviation from the diagonal of the curve (dots line) corresponding to points along the actual synchronous (solid line). For the parent system, the C2-C3 bond is suggested to form first and then the C1-C4 bond with asynchronicity degree of 0.11. The degree of asynchronicity of the substituted systems is shown in Table 1.
Table 1: The degree of asynchronicity, ∆d, at the transition states of intramolecular [2+2] cycloaddition reactions.
| C2-C3 | C1-C4 |
∆d |
|
|
TS1 |
2.21 | 2.16 | 0.06 |
| TS2 | 2.27 | 2.13 |
0.14 |
|
TS3 |
2.55 | 1.96 | 0.58 |
| TS4 | 2.22 | 2.12 |
0.10 |
|
TS5 |
2.38 | 2.01 | 0.37 |
| TS6 | 2.21 | 2.19 |
0.02 |
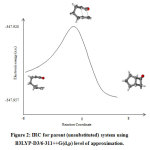 |
Figure 2: IRC for parent (unsubstituted) system using B3LYP-D3/6-311++G(d,p) level of approximation. Click here to view figure |
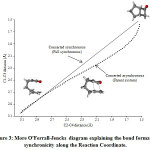 |
Figure 3: More O’Ferrall-Jencks diagram explaining the bond formation synchronicity along the Click here to view figure |
Reaction Coordinate.
The geometries of the transition states for a concerted mechanism involved in the intramolecular [2+2] cycloaddition reactions are given in Figure 4. The intramolecular reactions undergo cycloaddition through highly asynchronous transition states. An analysis of the lengths of the sigma forming bonds at the transition states indicates that the reaction follows asynchronous process. In all cases the length of the formed C1-C4 bonds are shorter than the formed C2-C3 bonds. The TS3 has larger ∆d among all transition states and TS6 has the smallest degree and the formation of the most favorable transition states is more asynchronous.
![Figure 4: Optimized geometries of the transition states involved in the intramolecular [2+2] cycloaddition reactions using B3LYP-D3/6-311++G(d,p) level of approximation.](https://www.orientjchem.org/wp-content/uploads/2019/10/Vol35_No5_The_Hay_fig4-150x150.jpg) |
Figure 4: Optimized geometries of the transition states involved in the intramolecular [2+2] cycloaddition reactions using B3LYP-D3/6-311++G(d,p) level of approximation. Click here to view figure |
The relative electronic energies of the transition states and their corresponding products in the gas phase are presented in Figure 5. As shown from these figures, the TS6 has higher energy than the other transition states. In contrast, TS3 has the lowest energy among all transition states. It is worth to realize that the P2 product is more stable than the others, due to their lowest electronic energies. From the observed data of activation energies and degree of asynchronicity, the transition states that have higher activation energies have the lowest degree of asynchronicity.
The thermodynamic parameters of the studied reactions in the gas phase at 1 atm and 298.15 K are collected in Table 2. The same trends of relative energies can be seen for enthalpy and Gibbs free energy values. An analysis of Table 1 indicates that the values of Gibbs free energy for products are negative, which refer to that the reactions are possible to occur. It is also shown that the transition states and products are found to be disfavored entropically. The activation enthalpy and activation Gibbs free energy at all levels of theory are found to be positive and very close. The overall cycloaddition reactions are exothermic and this indicates the stability of the products.
HOMO and LUMO energies in eV at B3LYP-D3/6-311++G(d,p) level of theory are computed for reactants and products and listed in Table 3. It is worth to realize that the energy gap for the parent molecules is higher than the other derivatives which indicates the stability of this product. Product P3 has narrow energy gap with 5.64 eV. The energy gap has become narrow with –NH2 and -CN (4.49 and 4.71, respectively). The wider energy gap was found for reactants R4 and R5.
![Figure 5: Energy profile for the intramolecular [2+2] cycloaddition reactions using B3LYP-D3/6-311++G(d,p) level of approximation. The energies are given relative to reactants (kcal/mol).](https://www.orientjchem.org/wp-content/uploads/2019/10/Vol35_No5_The_Hay_fig5-150x150.jpg) |
Figure 5: Energy profile for the intramolecular [2+2] cycloaddition reactions using B3LYP-D3/6-311++G(d,p) level of approximation. The energies are given relative to reactants (kcal/mol). Click here to view figure |
Table 2: Thermodynamic parameters for transition states and products at B3LYP-D3/6-311++G(d,p) level of theory in (kcal/mole) for ΔH and ΔG and in (cal/mol.K) for ΔS.
|
∆H (kcal/mol) |
∆G (kcal/mol) |
∆S (cal/mol.K) |
|
|
P1 |
-14.10 | -10.31 | -12.71 |
| P2 | -15.28 | -11.25 |
-13.50 |
|
P3 |
-8.15 | -4.62 | -11.84 |
| P4 | -13.01 | -9.39 |
-12.16 |
|
P5 |
-8.77 | -5.39 | -11.33 |
| P6 | -12.05 | -8.52 |
-11.85 |
|
TS1 |
46.94 | 50.72 | -12.69 |
| TS2 | 46.29 | 50.24 |
-13.25 |
|
TS3 |
39.59 | 43.19 | -12.07 |
| TS4 | 47.51 | 50.97 |
-11.60 |
|
TS5 |
45.42 | 48.96 | -11.88 |
| TS6 | 51.07 | 54.42 |
-11.26 |
Table 3: HOMO energies, LUMO energies and energy gap (in eV unit) for reactants and products at B3LYP-D3/6-311++G(d,p) level of theory.
|
EH |
EL |
EL-EH |
|
|
R1 |
-6.45 | -1.28 | -5.17 |
| R2 | -6.41 | -1.27 |
-5.14 |
|
R3 |
-5.75 | -1.30 | -4.45 |
| R4 | -6.43 | -1.16 |
-5.27 |
|
R5 |
-6.27 | -1.03 | -5.25 |
| R6 | -6.50 | -1.90 |
-4.60 |
|
P1 |
-6.83 | -1.05 | -5.78 |
| P2 | -6.78 | -0.95 |
-5.83 |
|
P3 |
-6.59 | -1.02 | -5.57 |
| P4 | -7.11 | -1.32 |
-5.79 |
|
P5 |
-6.87 | -1.07 | -5.80 |
| P6 | -7.45 | -1.58 |
-5.88 |
A good explanation for the regioselectivity in intramolecular [2+2] cycloaddition reactions may be obtained by calculating Fukui functions, where the transition structure associated with the rate-determining steps mostly involves the formation of one single bond between the most nucleophilic and other electrophilic sites in the [2+2] cycloaddition reagent pair. The global and local properties of reactants involved in the intramolecular reactions are gathered in Table 4. The nucleophilic site in all reactants are at C2 atom, which has the highest value of the Fukui function for an electrophilic attack , except R3 in which C4 atom has the highest value. The highest value of local electrophilicity (ωk) is located at C1 carbon atom, except for R6 where C4 has the highest value. The C4 carbon atom in R6 is considered the most powerful electrophile as the most favorable site for nucleophilic attack based on the local electrophilicity, ωk =0.635 eV, and the Fukui function, ![]()
Table 4: The Global and Local properties of reactants involved in intramolecular [2+2] cycloaddition reactions.
|
ω |
N | site (k) | ωk |
Nk |
|||
|
R1 |
1.45 |
3.04 |
C1 |
0.1195 | 0.3306 | 0.389 | 0.322 |
| C2 | 0.472 | 0.0477 | 0.056 |
1.273 |
|||
|
C3 |
0.0016 | 0.0019 | 0.002 | 0.004 | |||
| C4 | 0.0004 | 0.0042 | 0.005 |
0.001 |
|||
|
R2 |
1.43 |
3.08 |
C1 |
0.0933 | 0.2984 | 0.351 | 0.254 |
| C2 | 0.3683 | 0.0429 | 0.050 |
1.004 |
|||
|
C3 |
0.0127 | 0.0015 | 0.002 | 0.035 | |||
| C4 | 0.024 | 0.0006 | 0.001 |
0.065 |
|||
|
R3 |
1.40 |
3.74 |
C1 |
0.0028 | 0.3443 | 0.380 | 0.010 |
| C2 | 0.0069 | 0.0461 | 0.051 |
0.023 |
|||
|
C3 |
0.1247 | 0.0004 | 0.001 | 0.423 | |||
| C4 | 0.4054 | 0.0012 | 0.001 |
1.377 |
|||
|
R4 |
1.37 |
3.06 |
C1 |
0.1164 | 0.3717 | 0.416 | 0.317 |
| C2 | 0.4631 | 0.0304 | 0.034 |
1.259 |
|||
|
C3 |
0.0022 | 0.0021 | 0.002 | 0.006 | |||
| C4 | 0.0035 | 0.0019 | 0.002 |
0.010 |
|||
|
R5 |
1.27 |
3.21 |
C1 |
0.1155 | 0.3747 | 0.386 | 0.332 |
| C2 | 0.4476 | 0.0285 | 0.029 |
1.285 |
|||
|
C3 |
0.0082 | 0.0014 | 0.001 | 0.024 | |||
| C4 | 0.02 | 0.0011 | 0.001 |
0.057 |
|||
|
R6 |
1.92 |
2.99 |
C1 |
0.1148 | 0.0083 | 0.013 | 0.303 |
| C2 | 0.4673 | 0.002 | 0.003 |
1.233 |
|||
|
C3 |
0.0014 | 0.2547 | 0.389 | 0.004 | |||
| C4 | 0.0019 | 0.4158 | 0.635 |
0.005 |
Conclusions
The B3LYP-D3/6-311++G(d,p) level of approximation was used to investigate the concerted mechanism of the intramolecular [2+2] cycloaddition reaction of ketenes to obtain cyclobutanones as a cycloadducts. Different substitution groups were used to study their effect on the stability of the reaction products. The highest activation energy was found with CN group and lowest with NH2 group. All cycloadducts were found to be stable thermodynamically. The global and local properties including Fukui functions were studied for carbon centers of each reactant.
Acknowledgment
We would like to acknowledge the Department of Chemistry in University of Zakho and Department of Education in Salahaddin University.
Conflict of Interest
We as authors declare that there are no conflicts of interest with any person and organization related to this article.
References
- Rigsbee, E. M.; Zhou, C.; Rasik, C. M.; Spitz, A. Z.; Nichols, A. J.; Brown, M. K., Lewis acid-promoted [2 + 2] cycloadditions of alkenes with aryl ketenes. Organic & Biomolecular Chemistry 2016, 14 (24), 5477-5480.
- De Mesmaeker, A.; Kolleth, A.; Dagoneau, D.; Quinodoz, P.; Lumbroso, A.; Avanthay, M.; Catak, S.; Sulzer-Mossé, S., Synthesis of benzazepinones via intramolecular cyclization involving ketene iminium intermediates. Helvetica Chimica Acta 2019, 0 (ja).
- Lumbroso, A.; Catak, S.; Sulzer-Mossé, S.; De Mesmaeker, A., Efficient access to functionalized cyclobutanone derivatives using cyclobuteniminium salts as highly reactive Michael acceptors. Tetrahedron Letters 2015, 56 (19), 2397-2401.
- Neyyappadath, R. M.; Greenhalgh, M. D.; Cordes, D. B.; Slawin, A. M. Z.; Smith, A. D., Synthesis of Fused Indoline-Cyclobutanone Derivatives via an Intramolecular [2+2] Cycloaddition. European Journal of Organic Chemistry 2019, 0 (0).
- Tran, V.; Minehan, T. G., Intramolecular [2+ 2] Cycloaddition Reactions of Alkynyl Ether Derived Ketenes. A Convenient Synthesis of Donor–Acceptor Cyclobutanes. Organic letters 2011, 13 (24), 6588-6591.
- Lachia, M.; Jung, P. M. J.; De Mesmaeker, A., A novel approach toward the synthesis of strigolactones through intramolecular [2+2] cycloaddition of ketenes and ketene-iminiums to olefins. Application to the asymmetric synthesis of GR-24. Tetrahedron Letters 2012, 53 (34), 4514-4517.
- Lee-Ruff, E., Synthesis of Cyclobutanes. 2009; Vol. 38.
- Snider, B. B., Intramolecular cycloaddition reactions of ketenes and keteniminium salts with alkenes. Chemical Reviews 1988, 88 (5), 793-811.
- Snider, B. B.; Walner, M., Preparation of unsaturated α,α-dichloro acid chlorides and intramolecular [2 + 2] cycloadditions of the α-chloroketenes reductively generated from them. Effect of double bond geometry on the cycloaddition. Tetrahedron 1989, 45 (10), 3171-3182.
- Coquerel, Y.; Rodriguez, J., The Wolff Rearrangement: Tactics, Strategies and Recent Applications in Organic Synthesis. Molecular Rearrangements in Organic Synthesis 2015, 59-84.
- Zhang, L.; Dong, J.; Xu, X.; Liu, Q., Chemistry of Ketene N,S-Acetals: An Overview. Chemical Reviews 2016, 116 (2), 287-322.
- Souillart, L.; Cramer, N., Regiodivergent Cyclobutanone Cleavage: Switching Selectivity with Different Lewis Acids. Chemistry – A European Journal 2015, 21 (5), 1863-1867.
- Smit, A.; Kok, J. G. J.; Geluk, H. W., Thermal isomerization of β-ionylideneacetaldehyde. Isolation of a tricyclo[5,4,0,01,5]undec-3-en-6-one. Journal of the Chemical Society, Chemical Communications 1975, (13), 513-514.
- Hanford, W.; Sauer, J. C., Preparation of ketenes and ketene dimers. Organic reactions 2004, 3, 108-140.
- Charette, A. B., Handbook of Reagents for Organic Synthesis: Reagents for Heteroarene Functionalization. Wiley: 2015.
- de Meijere, A.; Bushby, R. J.; Eilbracht, P.; de Kimpe, N.; Goldschmidt, Z., Houben-Weyl Methods of Organic Chemistry Vol. E 17b, 4th Edition Supplement: Carbocyclic Three-Membered Ring Compounds, Cyclopropanes: Synthesis. Thieme: 2014.
- Marko, I.; Ronsmans, B.; Hesbain-Frisque, A. M.; Dumas, S.; Ghosez, L.; Ernst, B.; Greuter, H., Intramolecular [2+2] cycloadditions of ketenes and keteniminium salts to olefins. Journal of the American Chemical Society 1985, 107 (7), 2192-2194.
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 09 B.01, Wallingford, CT, 2009.
- Parr, R. G.; Weitao, Y., Density-Functional Theory of Atoms and Molecules. Oxford University Press: 1989.
- Khabashesku, V. N.; Kudin, K. N.; Margrave, J. L., Density functional theoretical studies of [2+2] cycloaddition of simple transient silenes and germenes to ethylene, formaldehyde, and thioformaldehyde, and vibrational analysis of spectra of reactants and cyclic products. Russian Chemical Bulletin 2001, 50 (1), 20-28.
- Lemal, D. M., Pathways for Concerted [2 + 2] Cycloaddition to Cumulenes. The Journal of Organic Chemistry 2017, 82 (24), 13012-13019.
- Ditchfield, R.; Hehre, W. J.; Pople, J. A., Self‐Consistent Molecular‐Orbital Methods. IX. An Extended Gaussian‐Type Basis for Molecular‐Orbital Studies of Organic Molecules. 1971, 54 (2), 724-728.
- Lee, C.; Yang, W.; Parr, R. G., Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B 1988, 37 (2), 785-789.
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. The Journal of Chemical Physics 2010, 132 (15), 154104.
- Fukui, K., Formulation of the reaction coordinate. The Journal of Physical Chemistry 1970, 74 (23), 4161-4163.
- Legault, C., CYLview 1.0. 2009.
- Parr, R. G.; Szentpály, L. v.; Liu, S., Electrophilicity Index. Journal of the American Chemical Society 1999, 121 (9), 1922-1924.
- Parr, R. G.; Pearson, R. G., Absolute hardness: companion parameter to absolute electronegativity. Journal of the American Chemical Society 1983, 105 (26), 7512-7516.
- Parr, R. G.; Weitao, Y., Density-Functional Theory of Atoms and Molecules. Oxford University Press: 1994.
- Kohn, W.; Sham, L. J., Self-Consistent Equations Including Exchange and Correlation Effects. Physical Review 1965, 140 (4A), A1133-A1138.
- Domingo, L. R.; Chamorro, E.; Pérez, P., Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. The Journal of Organic Chemistry 2008, 73 (12), 4615-4624.
- Domingo, L. R.; Aurell, M. J.; Pérez, P.; Contreras, R., Quantitative Characterization of the Local Electrophilicity of Organic Molecules. Understanding the Regioselectivity on Diels−Alder Reactions. The Journal of Physical Chemistry A 2002, 106 (29), 6871-6875.
Accepted on: 20-10-2019







ISSN Online: 2231-5039

![]()




{kind=link}