Synthesis of Oxadiazole Derivative of Pentacyclic Triterpenoid and Its Biological Activity
Mohammed Golam Rasul*
Qaseem Private Colleges, Department of Basic Medical Sciences, Post Doc Research fellow, Raiganj University, West Bengal, India.
Corresponding Author E-mail: golamrasul2008@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/350331
Article Received on : 07-02-2019
Article Accepted on : 02-05-2019
Article Published : 03 May 2019
Triterpenoid betunilic acid is extracted from outer bark of Biscofia javanica blume from Darjeeling hilly region and carried out transformative reaction to introduce oxadiazole moiety to ring A of the triterpenoid which was identified as 28-carbomethoxy lupan (2,3-c)-1′,2′,5′-oxadiazole. The derivative obtained has been selected for its antibacterial and fungicidal activity at different concentrations with respect to the parent compound. The structures of this compound were established based on spectroscopic (UV, IR, NMR) analysis.
KEYWORDS:Biological Activity; Extraction; Phytotoxicity; Transformative Reaction; Triterpenoid
Download this article as:| Copy the following to cite this article: Rasul M. G. Synthesis of Oxadiazole Derivative of Pentacyclic Triterpenoid and Its Biological Activity. Orient J Chem 2019;35(3). |
| Copy the following to cite this URL: Rasul M. G. Synthesis of Oxadiazole Derivative of Pentacyclic Triterpenoid and Its Biological Activity. Orient J Chem 2019;35(3). Available from: https://bit.ly/2ZTVmYn |
Introduction
Plant materials are on increasing interest for their applications in pharmaceutical, nutritional and cosmetic applications. They represent a source of active ingredients known for long times ago by its traditional uses for medical purposes. Plants are rich in active compounds or secondary metabolites such as alkaloids, terpenoids, steroids, glycosides, saponins, flavonoids and polyphenols, which are present in their organs such as leaves, flowers, bark, seeds, fruits etc. In recent years, the medicinal values of pentacyclic triterpenoids have been increasingly recognized and studied because of their anti-inflammatory,1 hypoglycemic,2 antibacterial,3 antioxidant,4 antitumor,5 anti proliferative,6 anti-HIV, anti-cancer,7 antitrypanosomal,8 antileishmanial,9 anti-arthritic,10 hepatoprotective,11 cardiovascular,12 and immunological adjuvant13 activities. In the past decade, numerous reports indicated that pentacyclic triterpenoids exhibit various bioactivities. Among these triterpenoids, betulinic acid is of special interest due to its significant biological activity exhibited both by the acid itself and by its numerous derivatives.14 A number of derivatives of it have also been prepared either to establish the structure of the compounds or to study the reactivity of such compounds towards some specific oxidizing and reducing agents.15,16 Synthetic transformations of natural compounds for the purpose of developing biological activity have become the basis of the scientific direction of perfect organic synthesis and natural product chemistry. The author is interested in the biological activity of the pentacyclic terpenoid betulinic acid and its derivativeto understand its mode of action, determining structure activity relationship (SAR) and making suitable derivatives with improved biocidal activity.
Materials and Method
Isolation of the Compound from the barks of Xanthoxylum Budrunga
First collected the bark of Biscofia javanica blume from Darjeeling hilly region, dried on sunlight and coarsely powdered. These powdered mass were extracted with toluene in a soxhlet apparatus for 36 hours. The solvents were then removed under reduced pressure and a sticky brown residue was obtained. This crude toluene extracts of the plant were then purified over a column chromatography using silica gel of 60-120 mesh using petroleum ether (PE) and ethyl acetate (EA) mixture as an eluent with increasing concentration.
Elucidation of the Structures
All the melting points were determined by open capillary method and are uncorrected. The NMR spectra were recorded in CDCl3 solutions at ambient temperature on a Bruker Avance 300 MHz-FT NMR spectrometer using 5 mm BBO probe. The chemical shift d values are given in ppm related to tetra methyl silane (TMS) as internal standard. The coupling constants (J) are reported in Hz. The IR spectra were recorded in Shimadzu FT-IR spectrophotometer in KBr discs.
Characterization of the Isolated Compound as Betulinic Acid 1
Different fractions of isolated compounds were mixed and crystallized by chloroform and methanol mixture which gave white crystals m.p 299-301°C. IR spectrum has exhibited hydroxyl at vmax 3610, 1020 cm-1 and exomethylene at vmax 3060, 1630, 880 cm-1. The 1H NMR spectrum revealed signals for five tertiary methyl δH 0.65, 0.75, 0.90, 0.96 and 0.98, a vinyl methyl δH = 1.97 (broad d, J = 0.5 Hz), a secondary carbinol δH = 3.16 (dd, J = 9.5 and 6.0 Hz) and δH = 2.95 (dd, J = 9.0, 6.0 and 0.5 Hz) an exomethylene group δH = 4.55 (1H, d, J = 0.4 Hz) and δH = 4.65(1H, d, J = 0.4 Hz). These data indicated a pentacyclic triterpenoid of betulinic acid, confirmed by comparison with already published data.17 The 13C NMR spectrum showed six methyl group at δC 27.9 (C-23), 15.4 (C-24), 16.2 (C-25), 16.3 (C-26), 14.6 (C-27), 19.6 (C-30) and exomethylene group at δC 150.0 (C-20), 108.8 (C-29) and a secondary carbon bearing hydroxyl at dc 79.0 (C-3) and a carboxyl group at δC = 180.6 (C-28) in addition to ten primary carbon atom, five secondary carbon atom and five tertiary carbon atom. These data were identical to those five secondary carbon atom and five tertiary carbon atom. These data were identical to those reported for betulinic acid.18
Preparation of Dihydromethylbetulinate 2
Betulinic acid is treated with diazomethane in ether and kept it overnight and the product obtained dissolved in ethyl acetate was shaken in an atmosphere of hydrogen in presence of palladium in charcoal catalyst for three hours until absorption of hydrogen ceased. Crystallization from a mixture of chloroform and methanol furnished colorless needle shaped compound m.p. 235—237°C, [a]D ─17.0°C. IR spectrum showed peaks at 3540 cm-1 (OH) and 1705(C=O) cm-1. This compound was found to be identical with an authentic sample of dihydromethylbetulinate 2 (m.m.p, CO TLC, CO IR).
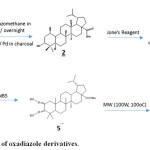 |
Scheme 1: Preparation of oxadiazole derivatives. |
Preparation of dihydromethylbetulonate 3
To a solution of 2 (2.95 g) in pure acetone added Jone’s reagent drop wise with constant shaking until a faint orange color persisted. The resulted product was chromatographed which developed with petroleum ether and elution with pet. ether: ethyl acetate (90: 10 v/v) furnished different fractions of a single compound. The different fractions were mixed and on crystallization from methanol furnished needle shaped crystals of dihydromethylbetulonate 3, m.p. 190—192°C, [a]D + 8.2°. IR spectrum showed peaks at 1730 cm-1 (─COOMe) and 1708 cm-1 (C=O) identical with an authentic sample of dihydromethylbetulonate (m.m.p, CO TLC, CO IR) [Lit m.p. 194oC, [a]D + 8.4°].
Preparation of 2, 2-dibromodihydromethylbetulonate 4
A solution of 3 in chloroform was mixed with dimethylsulphoxide. N-Bromosuccinimide (NBS) was then added to the solution with constant shaking.So the residue was chromatographed over silica gel column. The chromatogram was developed with petroleum ether and elution with pet. ether: ethyl acetate (90:10 v/v) furnished a single compound, by spectral analysis the compound is identified as 2, 2-dibromodihydromethyl betulonate 4.
Preparation of 28-carbomethoxy-2, 3-dioximinolupane 5
Compound 4 was dissolved in pyridine and refluxed with hydroxyl amine hydrochloride in ethanol. The compound obtained from the reaction was purified by repeated crystallization from chloroform-methanol mixture to obtain a white amorphous powder of compound A, analyzed for C32H53O4N2, IR spectrum showed peaks at 3200-3400 cm-1 (O-H) 1720 cm−1 (−COOMe). It exhibited UV absorption maximum at 220 nm (€=5100). Mass spectrum of the compound showed molecular ion peak at m/z 529 [M]+ the other peaks at prominence appearance at m/z 530, 531 (base peak), 424, 422, 380, 341, 340, 299, 231, 191, 163, 149, 136, 122, 121, 95, 81, 69. PMR spectrum of A could not be taken owing to solubility problem. Thus on the basis of the above spectral data compound A was identified as 28-carbomethoxy-2,3-dioximinolupane 5.
Preparation of 28-carbomethoxy lupan [2,3-c]-1/,2/,5/-oxadiazole 6
28-carbomethoxy lupan [2,3-c]-1’,2’, 5’-oxadiazole 6 was prepared by cyclization of 5 in dry DMF under microwave irradiation (100W, 100oC) for 10 minutes. The residue obtained after usual work up was purified by repeated crystallization from chloroform-methanol mixture and designated compound 6, analyzed for C31H48O4N2.Which was corroborated by mass spectral analysis that showed the molecular ion at m/z 496. The other ions appeared at m/z 496.72 [M-CH3] +, 496 (base peak), 497, 367, 271, 259, 245, 231, 206, 191, 163, 149, 123, 121, 109, 95, 81, 55. IR spectrum of the compound showed peaks at 1620 cm-1 (─C=N-O) and 890 cm-1, 1720 cm−1 (−COOMe) for heterocyclic ring. It showed UV absorption maximum at 223 nm (€=5169) for disubstituted furazan derivative. The 1HNMR are 0.77 (d, 3H, J=7 Hz) 0.78, 0.86 (d, 3H, J=7 Hz), 0.97, 1.10. It showed the presence of six tertiary methyl. A pair of one-proton doublets each at 2.1 (J=16 Hz) ppm and 3.15 (J= 16 Hz) ppm may be due to the geminal coupling of the C-1 proton which are adjacent to the furazan ring. Thus from spectral analysis the structure of the compound 13 has been established as 28-carbomethoxy lupan [2,3-c]-1’, 2’, 5’-oxadiazole.
Assay of Antibacterial Activity by the Disc Diffusion Method
One ml of 48 h old culture of the test bacterium was taken in a Petri dish of 90 mm diameter. Then 20 ml of sterile nutrient agar (NA) medium was poured in the Petri dish and shaken carefully to mix the bacterial suspension with the medium. The Petri dishes were allowed to cool. Filter paper discs of 5 mm diameter containing desired concentration of the test samples were placed on the surface of the solidified media and incubated at 37oC in an incubator for 48 h. Diameter of the inhibition zones were measured and reported in Table 1. In control sets, no chemicals were used in the filter paper discs but sterile distilled DMSO was used to soak the filter papers. The antibacterial activities were compared with ampicillin, a b-lactam antibiotic.
Assay of Antifungal Activity
Fungi were grown on potato dextrose agar (PDA) medium at 28 ± 1oC for mycelial growth. The fungicidal activities were determined using agar cup bioassay and spore germination bioassay. The purified eluents (10 ml) were placed on two spots 3 cm apart on a clean grease free slide and the solvent was allowed to keep for some time to evaporate. One drop (0.02 ml) of spore suspension (10spores/ml) prepared from 15 days old culture of the test fungi was added on the same place where the purified eluents were placed and subsequently evaporated. Compound 1 and 6 dissolved in DMSO (dimethyl sulfoxide) and five different concentrations were prepared (500 ppm, 400 ppm, 300ppm, 200 ppm, 100 ppm). The studies were performed at 28±1oC for 24 h under humid conditions in Petri plates and reported in Table 2. Finally, after proper incubation period, one drop of a cotton blue-lacto phenol mixture were added to each spot to fix the germinated spores. The number of spores germinated were compared with that of germinated spores of control (where no chemicals were used). Moist chamber were used for germination of spores. The number of germinated spores were calculated based on an average of 300 spores per treatment. The antifungal activities were compared with streptomycin.
Result and Discussion
Antibacterial Activity
Two different compounds oxadiazole derivative of 28-carbomethoxy lupan [2,3-C]-1’, 2’, 5’-oxadiazole 6 and the parent compound betulinic acid 1 were tested for their antibacterial properties against five different bacteria E. coli, B. subtilis, S. aureus, L. salivarius and P. aeruginosa.
Table 1: Results of antibacterial activity.
| Microorganism | Compounds under Investigation | Inhibition zone (cm) | ||
| 100 ppm | 250 ppm | 500 ppm | ||
| E. coli ( MTCC-739) | 6/1 | 2.6/ 2.3 | 2.7/ 2.4 | 2.8/ 2.5 |
| B. subtilis (Identified by NBU) | 6/1 | 1.8/ 1.6 | 1.9/ 1.8 | 2.4/ 2.3 |
| S. aureus (MTCC-26) | 6/1 | 1.7/ 1.6 | 1.9/ 1.8 | 1.9/ 1.8 |
| L. salivarius (Identified by NBU) | 6/1 | 2.4/ 1.9 | 2.3/ 2.0 | 2.4/ 2.3 |
| P. aeruginosa (MTCC-2453) | 6/1 | 1.2/ 1.1 | N.P/0.9 | 1.2/ 1.0 |
NP= Not prominent, 1=Betulinic acid, 6= 28-carbomethoxy lupan [2,3-c]-1’, 2’, 5’-oxadiazole.
From the result it was evident that significant inhibition of growth were observed both in the case of E. coli and Lactobacillus by all the concentrations of oxadiazole derivative of 28-carbomethoxy lupan [2,3-C]-1’, 2’, 5’-oxadiazole 6 with respect to parent compound Betulinic acid 1 tested in disc diffusion method. For oxadiazole derivative 6 maximum inhibition was observed at 500 ppm when tested on E. coli; the inhibition zone was 2.8 cm, but at 250 ppm and 100 ppm concentration the diameter of inhibition zones were 2.7 cm and 2.6 cm respectively. Thus there is a gradual increase in the zone of inhibition with the increase in concentration of the compounds. Similar results were also observed in case of the parent compound 1 at 500 ppm, 250 ppm and 100 ppm concentrations the diameters of inhibition zones were 2.5 cm, 2.4 cm and 2.3 cm respectively. Although the inhibition values were found relatively less with B. subtilis, S. aureus in comparison to E. coli and Lactobacillus but the trends of activity were found identical. The growth of inhibition zone with Pseudomonas was less compared to other microorganisms for both 6 and 1 and at 250 ppm for 6 it was not prominent.
Antifungal Activity
The antifungal activities of betulinic acid 1 and 28-carbomethoxy lupan [2,3-c]-1’, 2’, 5’-oxadiazole 6 at different concentrations were tested against F. solani.
Table 2: Results of Antifungal Activity.
| Compounds | Concentration | % of germination | % of inhibition | Range of germ tube length |
| Control | 97.77 | 0 | 3.0-5.0 | |
| 100 ppm | 12.4/83.86 | 86.65 /13. | 2.4-4.9 / 2.6-5.9 | |
| 6/1 | 200 ppm | 8.20/17.75 | 91.34/81.79 | 2.2-4.6 / 2.3-4.5 |
| 300 ppm | 5.10/16.24 | 94.51 /83.51 | 2.3-4.5 / 2.4-4.5 | |
| 400 ppm | 2.56/9.54 | 97.39 /91.94 | 1.7-3.8 / 2.3-4.2 | |
| 500 ppm | 1.00/4.44 | 98.72 / 96.28 | 1.5.34 / 2.3-4.0 |
1=betulinic acid, 6= 28-carbomethoxy lupan [2,3-c]-1’, 2’, 5’-oxadiazole
The parent compound betulinic acid 1 and the respective oxadiazole derivative 28-carbomethoxy lupan [2,3-c]-1’, 2’, 5’-oxadiazole 6 showed inhibition of spore germination and inhibitory effect on the growth of F. solani. Parent compound 1 at 100 ppm concentration showed 83.88% germination and only 12.40% germination where as 97.77% germination was experienced in control set. Similar results were also observed with oxadiazole derivative 6 at the same concentrations. However, at 500 ppm concentration both betulinic acid 1 and oxadiazole derivative 6 significantly reduced germination of spores. It showed 96.82 % and 98.72% inhibition of spore germination whereas in control set there was no inhibition.19
Conclusion
The derivative 6 showed better activity than parent compound 1. The introduction of oxadiazole moiety to ring A of the pentacyclic triterpenoid enhanced the activity of the compound obtained from natural sources. The present study will be extremely helpful to enrich the present knowledge about the structure activity relationship for this type of triterpenoid skeleton and encouraged the author towards a biological potential on a library of pentacyclic triterpenoids and their derivatives.
Acknowledgements
I would like to express my deep sense of gratitude and thankfulness to my esteemed respected Supervisor Prof. Pranab Ghosh, Department of Chemistry, University of North Bengal and Authorities of Raiganj University for providing Laboratory facilities and their support.
Conflict of Interest
There is no conflict of interest.
References
- Yadav, V.R.; Prasad, S.; Sung, B; Kannappan, R.; Aggarwal, B.B. Toxins, 2010, 2, 2428–2466.
- Li, J.J.; Li, Y.; Bai, M; Tan, J.F.; Wang, Q.; Yang, J. Biomed. Chromatogr., 2014, 28, 717–724.
- Penduka, D.; Mosa, R.; Simelane, M.; Basson, A.; Okoh, A.; Opoku, A. Ann. Clin. Microbiol. Antimicrob., 2014, 13, 37.
- Kim, Y.A.; Kong, C. S.; Lee, J. I.; Kim, H.; Park, H.Y.; Lee, H.S.; Lee, C.; Seo, Y. Bioorg. Med. Chem. Lett., 2012, 22, 4318–4322.
- Shanmugam, M.K.; Dai, X.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Cancer Lett., 2014, 346, 206–216.
- Memon, A.H.; Ismail, Z.; Al-Suede, F. S.; Aisha, A.F.; Hamil, M.S.; Saeed, M. A.; Laghari, M.; Majid, A. M. Molecules, 2015, 20,14212– 14233.
- Wang, H.; Zhang, C.; Wu, Y.; Ai, Y.; Lee, D.Y.; Dai, R. Biomed. Chromatogr. 2014, 28, 1402–1408.
- Jiangm, Y.; Liu, F.; Wang, Y.; Li, H. J. Pharm. Biomed. Anal. 2016, 132, 148–155.
- Quan, K.; Park, H. Y.; Oh, J.; Park, H.Y.; Ferreira, D. A.; Myung, C.; Na, M. J. Nat. Prod. 2016, 79, 2559–2569.
- Simao Juca Cruz , Md. F.; Pereira, G. M; Ribeiro, M. G.; DaSilva, A.M.; Tinoco, L.W.; da Silva, B.P.; Parente, J. P. Carbohydr. Res. 2016, 420, 23–31.
- Ghosh, P.; Rasul, M.G.; Chakraborthy, M.; Mandal, A.; Saha, A. Indian J. Chem. 2011, 50B, 1519-1523.
- Yadav, M.; Abraham, J.; Jindal, D.P. Indian J. Chem. 1990, 29B, 26-28.
- Rasul, M. G.; Ghosh, P.; Chakraborthy, M. IACS, 2005, 364.
- Pettit, G.R.; Numata, A.; Cragg, G. M.; Herald, D. L.; Takada, T.; Iwamoto, C. J Nat Prod. 2000, 63, 72–5.
- Peng, C.; Bodenhausen, G.; Qiu, S. X.; Fong, H. H. S.; Farnsworth, N. R.; Yuan, S. G.; Zheng, C. Magn Reson Chem. 1998, 36 , 267–78.
- Takeoka, G.; Dao L.; Taranishi, R.; Wong, R.; Flessa, S.; Harden, L.; Edwards, R. J. Agric. Food Chem. 2000, 48(8), 3437-3439.
- Mwangi, E. S. K.; Keriko, J. M.; Machocho, A. K.; Wanyonyi, A. W.; Malebo, H. M., J. Med. Plants Res. 2010, 4, 726–731.
- Ibrahim, S. R. M.; Abdallah, H. M.; Mohamed, G. A.; Ross, S. A. Fitoterapia, 2016, 112, 161–167.
- Shai, L.J.; Mc Gaw, L.J,; Aderoqba, M.A.; Mdee, L.K.; Eloff, J.N. J Ethnopharmacol. 2008,119, 238-44.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


