Synthesis, Characterization and Cytotoxic Activity of some new 1,2,3-Triazole, Oxadiazole and Aza- β-lactam Derivatives
Kany A. Abdulqader1, Ahmed W. Naser1 , Muthanna S. Farhan2 and Sabah J. Salih2
, Muthanna S. Farhan2 and Sabah J. Salih2
1Department of Chemistry, College of Science, University of Baghdad, Baghdad, Iraq.
2Department of Pharmaceutical Chemistry, College of Pharmacy, University of Baghdad, Baghdad, Iraq.
Corresponding Author E-mail address: kaneeazad@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/340516
Article Received on : 08-04-2018
Article Accepted on : 10-08-2018
Article Published : 10 Sep 2018
A series of 1,2,3-triazole, oxadiazole and aza-β-lactam derivatives were synthesized through consecutive reaction began from o-(N-propargyl) sulfonamido benzoic acid (1a). The reaction of (1a) with absolute ethanol in the presence of concentrated H2SO4 resulted in the formation of ester derivative (2a). The product of the previous reaction was reacted with 80% hydrazine hydrate to prepare benzohydrazide derivative (3a). 1,3,4-oxadiazole compound (4a) was obtained by condensation of compound (3a) with CS2 in presence KOH . Compound (3a) react with Phenyl isocyanates to give Carboxamide derivative (5a), that Condensation either with 2,4-dimethoxybenzaldhyde and p-hydroxybenzaldehyde to prepare the Schiff bases (6a-b). The cycloaddotion of Schiff-bases (6a-b) with phenyl isocyanate gave aza-β-lactams (7a-b). Benzamide derivatives (8a-c) were prepared via the reaction of compound (1a) with aniline derivatives, such as (p-toluidine, o-nitroaniline and m-nitroaniline). In a regioselective reaction 1,4-disubstituted-1,2,3-triazole derivative (9a-j) were synthesized via the click reaction of compounds 4a,5a and (8a-c) with benzyl azide and p-bromobenzyl azide. The compounds were identified using the spectral methods shown in the work. Cytotoxic effects of some final prepared compounds were studied in one cultured cellular models (MCF7 cell line) breast cancer (at various concentrations) by MTT assay, compound (9j) showed the better cytotoxic activity among the tested compounds.
KEYWORDS:Aza-β-lactam; Click Chemistry; Cytotoxicity; Oxadiazole; 1,2,3-Triazole
Download this article as:| Copy the following to cite this article: Abdulqader K. A, Naser A. W, Farhan M. S, Salih S. J. Synthesis, Characterization and Cytotoxic Activity of some new 1,2,3-Triazole, Oxadiazole and Aza- β-lactam Derivatives. Orient J Chem 2018;34(5). |
| Copy the following to cite this URL: Abdulqader K. A, Naser A. W, Farhan M. S, Salih S. J. Synthesis, Characterization and Cytotoxic Activity of some new 1,2,3-Triazole, Oxadiazole and Aza- β-lactam Derivatives. Orient J Chem 2018;34(5). Available from: http://www.orientjchem.org/?p=49554 |
Introduction
A wide ranging of various heterocyclic compounds has been discovered to develop pharmaceutically essential molecule. Among them which have played a significant role in medical chemistry were oxadiazoles derivatives.1,2 Which has been commonly used as a privileged scaffold for producing different novel pharmaceutical drug such as: antitumor,3 anti-cancer agents4 and anti prostate,5 furthermore, oxadiazole derivatives which have thioamide group CNS. Its importance lies in eliminating the poisons in much of medicine used by human beings.6 Azetidinones are one of the heterocyclic derivatives, which are utilized as synthase for various biologically active compounds,7 as well as their recognition as antibacterial,8 anti-inflammatory9 and anti-hepatitis.10 The triazole derivatives classified as very important heterocycles compound, over past few years they have acquired in interest by introduce the “click chemistry concept”.11-12 In which this (process) has concentrate on reaction between high reactives partner that was providing access to structure which easy varied. Huisgen’s thermal-cycloaddition of azide-alkyne obtain triazole,13 which catalyze by Cu (I)14-18 scheme (1). These situations describe “click” concept perfectly. This obtained 1,4-disubstituted regioisomer of 1,2,3-triazole and facilitate reaction at lesser temperature. In which increasing number of applications of Click chemistry was now being used in various researches such organic chemistry, bioconjugation,19 drug discovery,20 polymers,21-22 and radiochemistry.23
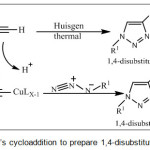 |
Scheme 1: Huisgen’s cycloaddition to prepare 1,4-disubstituted-1,2,3-triazoles. |
Experimental Methods
All chemicals were purchased from Fluka, BDH and Merck. M. p. is recorder use electrothermal (M. p.) apparataus. The FT-IR spectral data were recorded on a Shimadzu FT-IR8400S spectro-photometer in the Department of Chemistry, College of Science, University of Baghdad. 1H-NMR and 13C-NMR spectra are recorder on central laboratory of Isfahan University and Sharif University of technology, 400MHz, using CDCl3 or DMSO and tetramethylsilane (TMS) as an internal standard.
Synthesis of ethyl-o-(N-Propargyl) sulfoamido benzoate (2a)24
To a solution of o-(N-propargyl) sulfonamido benzoic acid (1a) (0.5gm, 0.004mole) in absolute ethanol (30ml), was added with stirring conc. sulfuric acid (2ml) the reaction mixture was refluxed for 8h, then the mixture was concentrated and neutralized by NaHCO3. The white precipitate that formed was filtered and re-crystallized from ethanol to give compound (2a).Yield 61%, M.p.(110-112oC), IR-KBr cm-1: 3274 (C-H) Acetylinic, 2125 (C C), 1737(C=O), 1056 (C-O) Ester. 1H-NMR (DMSO-d6) (δ ppm) 1.4(s, 3H, CH3), 2.45(s, 1H, CH acetylinic), 3.95(s, 2H, CH2), 4.55(O-CH2), 7.3.94(s, 1H, NH-C=O), 7.65-8.2(m, 4H, Ar-H).
Synthesis of o-(N-Propargyl) sulfoamido benzohydrazide (3a)25
Ethyl-o-(N-Propargyl) sulfoamido benzoate (2a) (0.5gm, 0.004 mole) was dissolved in (20ml) absolute ethanol, (0.05mole) of hydrazine hydrate was added and refluxed 4h. After cooling, white precipitate was formed and re-crystallized from EtOH to obtain compound (3a).Yield 70%, M.p.78-80 oC, IR-(KBr) cm-1: 3330-3242 NHNH2, 1656-1625(C=O) amide. 1H-NMR (DMSO-d6) (δ ppm) 1.45(s, 2H, NH2), 2.98(s, 1H, CH acetylinic), 3.91(s, 2H, CH2), 5.94(s, 1H, NH-C=O), 7.54-7.86 (m, 4H, Ar-H).
Synthesis of o-(5-mercapto-1,3,4-oxadiazol-2-yl)-N-propargyl benzene sulfonamide (4a)26
A mixture of Compound (3a) (0.5gm, 0.002 mole), with (0.004 mole) carbon disulfide and potassium hydroxide (0.2gm, 0.003 mole) was dissolved in (15ml) EtOH and refluxed 7h, then the solvent is evaporate and water was added into the residue until it dissolved and acidify with dilute (HCl), the precipitate is filtered, and washing with H2O to give compound (4a). Yield 83%, m.p 119-120oC, IR (KBr) cm-1: 3195(N-H), 2650(S-H), 1596 C=N. 1H-NMR (DMSO-d6) (δ ppm) 2.82(s, 1H, CH acetylinic), 3.45(s, 1H, NH-SO2), 3.8(s, 2H, CH2), 7.80-8.03(m, 4H, Ar-H).
Synthesis of N-Propargyl-o-[N\-sulfamido benzoyl] hydrazine carboxamide (5a)27
A mixture of o-(N-Propargyl) sulfoamido benzohydrazide (3a) (0.5gm, 0.002 mole) and Ph=C=O (0.002 mole) was refluxed in absolute ethanol (15ml) for 6h. After cooling the formed white precipitate was filtered, dried then re-crystallized from (1:1) EtOH and DMF, for giving compound (5a). Produce 85%, m.p 134-136oC, IR cm-1: 3298(N-H), 1672(C=O). 1H-NMR :3.09(s, 1H, CH acetylinic), 3.91(s, 2H, CH2), 7.4-8.33(m, 10H, Ar-H and SO2-NH), 9.30(s, 1H, NH).
General method to synthesis Schiff’s bases (6a-b)28
o-(N-Propargyl) sulfoamido benzohydrazide (3a) (0.5gm, 0.002mole) was dissolved In (20ml) absolute EtOH. Aromatic aldehydes namely 2,4-dimethoxybenzaldehyde and p-hydroxybenzaldehyde, (2-3) drops of glacial (acetic acid) were added and refluxed 5h. The forming precipitate after-cooling was filtered and re-crystallized from EtOH to give compounds (6a-b).
o-[(N-Propargyl)sulfamoyl]-N-(2,4-dimethoxybenzaldine)benzamide(6a)
Yielding 85%, m.p 160-163oC, IR cm-1: 3130(N-H), 1614(C=N), 1207 C-O. 1H-NMR (DMSO-d6) (δ ppm): 2.05(s, 1H, CH acetylinic), 3.7(s, 2H, CH2), 3.8(s, 6H, 2CH3-O), 5.97(s, 1H, NH-C=O), 6.54-8.46(m, 8H, Ar-H), 9(CH=N).
o-[(N-Propargyl)sulfamoyl]-N-(p-hydroxybenzaldine)benzamide(6b)
Yielding 73% m.p 212-214oC, IR-KBr cm-1: 3112(N-H), 1625(C=N), 3265 (OH). 1H-NMR: 2.1(s, 1H, CH acetylinic), 3.8(s, 2H, CH2), 6.04(s, 1H, NH-C=O), 7.3-8.1(m, 8H, Ar-H), 9.1(CH=N), 10(O-H).
General procedure for Synthesis of (Aza-β-lactam-derivative) (7a-b)29
A mixture of Schiff bases (7a-b) (0.5gm, 0.002 mole) and Ph=C=O (0.002 mole) in (15ml) chloroform was refluxed 6h. The Solvent was removed and the residue treated through a mixture of (1:1) ethyl acetate-petroleum ether. The resultant ppt. was filtrated then dried to give compounds (7a-b).
N-[2-oxo-3-phenyl-4-[(2,4-dimethoxy)phenyl-1,3-diazetidin-1-yl]-o-[(N-proparg yl) sulfamido] benzamide (7a)
Yield 85%, m.p 149-150oC, IR cm-1: 3328 (N-H), 1708 (C=O) Lactam, 1649 (C=O,Amide), 1232 (C-O).
N-[2-oxo-3-phenyl-4-[(p-hydroxy)phenyl-1,3-diazetidin-1-yl]-o-[(N-propargyl) sulfamido]benzamide (7b)
Yield 78%, m.p 161-162oC, IR cm-1: 3195 (N-H), 1712 (C=O) Lactam, 1649 (C=O)-Amide, 3315 (H-O).
General-method to synthesized of benzamide derivative (8a-c)30
Compound (1a) (0.5gm, 0.002 mole), with (0.002 mole) of aniline derivatives such as (p-toluidine, o-nitroaniline and m-nitroaniline) in 20 ml POCl3 was refluxed 4h. Then the mixture was Concentrated and poured on crushed ice with stirring for giving a solid mass. The product was filtered and dried to give compounds (8a-c).
o-[(N-Propargyl)sulfamoyl]-N-(p-tolyl)benzamide (8a)
Yielding 80%, m.p 99-101oC, IR cm-1: 3276(C-H)Acetylinic, 2130 (C C), 1735(C=O), 1H-NMR: 2.12(s, 3H, CH3), 2.85(s, 1H, CH acetylinic), 3.44(s,1H,NH-SO2), 4.59(s, 2H, CH2), 6.5(s,1H,NH-C=O), 6.48-8.34(m, 8H, Ar-H ). 13C-NMR (δ ppm): 27.46(CH3), 75-76 (C C), 121-136 (C-Ar), 158 (C=O).
o-[(N-Propargyl)sulfamoyl]-N-(m-nitrophenyl)benzamide (8b)
Yielding 76%, m.p 90-91oC, IR cm-1: 3276(C-H) Acetylinic, 2127 (C C), 1737(C=O).
o-[(N-Propargyl)sulfamoyl]-N-(o-nitrophenyl)benzamide (8c)
Yield 74%, m.p 96-97oC, IR cm-1: 3274 (C-H) Acetylinic, 2125(C C), 1737(C=O).
Preparation of alkyl azide31
Sodium azide (0.015 mole) was added slowly to refluxed DMF (25ml), the reflux was continued until all the sodium azide dissolved then benzyl chloride and p-bromobenzyl bromide (0.0075 mole) was slowly added, the mixture then was heated overnight (temperature is fixed at 750C), after cooling (25ml) of distilled H2O was added, 25ml of diethyl ether was also added and the organic-layer was extracted (the addition of diethyl ether was repeated three times),then the combined organic layers were dried using magnesium sulfate and evaporated under reduced pressure. To give the colorless liquids, n-benzyl azide 80% and p-bromobenzyl azide 83%, IR (KBr) cm-1: 2096 (N3).
General procedure of synthesis 1,4-disubstituted-1,2,3-triazole derivatives (9a-j)31
A mixture of compounds 4a, 5a and (8a-c) (0.002 mole) was slowly added with stirring to a solution containing sodium ascorbate (0.00045 mole) and CuSO4.5H2O (0.002 mole) in (25ml) DMF, then p-bromobenzyl azide and benzyl azide (0.005 mole) was added. The mixture then heated overnight (the temperature was fixed at 75oC), after cooling 25ml of distilled water was added, 25ml of diethyl ether was also added to the mixture and the organic-layer was extracted (the addition of diethyl ether was repeated three times), then the combined organic-layers were dried over magnesium sulfate, the forming ppt. was filtered and dried to give compounds (9a-j).
o-(5-mercapto-1,3,4-oxadiazol-2-yl)-N-(1-benzyl-1,2,3-triazol-4-yl)methyl benzene sulfonamide (9a)
The product 80%, m.p 212-215 oC, IR cm-1: 3338(N-H), 1618(N=N), 1164(C=S). 1H-NMR: 4.99(s, 2H, CH2), 5.58(s, 2H, benzylic), 7.25-8.33(m, 13H, Ar-H and SO2-NH), , 11.77(s, 1H, SH). 13C-NMR (δ ppm): 31(NH-CH2), 52(C-benzylic), 127-133 (C-Ar).
o-(5-mercapto-1,3,4-oxadiazol-2-yl)-N-(1-(p-bromobenzyl-1,2,3-triazol-4-yl)) methyl benzene sulfonamide (9b)
The product 66%, m.p 175-176oC, IR (KBr) cm-1: 3423(N-H), 1564 (N=N), 1161(C=S).
o-[(N-1-benzyl-1,2,3-triazol-4-yl)]methyl sulfamido-N-phenyl benzoyl hydrazine carbamide (9c)
The product 65%, m.p 228-231oC, IR (KBr) cm-1: 3471(N-H), 1720(C=O), 1562 (N=N).
o-[(N-(1-(p-bromobenzyl))-1,2,3-triazol-4-yl)]methylsulfamido-N-phenyl benzoyl hydrazine carbamide (9d)
The product 78%, m.p 180-183oC, IR (KBr) cm-1: 3409(N-H), 1708 (C=O), 1596 (N=N), 757 (C-Br). 1H-NMR: 4.80(s, 2H, CH2), 5.41(s, 2H benzylic), 7.25-8.33(m, 15H, Ar-H and SO2-NH), 9.30 (s, 1H, NH). 13C-NMR (δ ppm): 45(NH-CH2), 60(C- benzylic), 121-138(C-Ar), 153-166(C=O).
o-[N-(1-benzyl)-1,2,3-triazol-4-yl]methylsulfamoyl-N-(p-tolyl)benzamide(9e)
Yielding 80% , m.p 198-199oC, IR-KBr cm-1: 3452 (N-H), 1726 (C=O), 1596 (N=N).
o-[N-(1-(p-bromobenzyl))-1,2,3-triazol-4-yl]methylsulfamoyl-N-(p-tolyl) benzamide (9f)
Yielding 75%, m.p 164-165oC, IR cm-1: 3422(N-H), 1724(C=O), 1595(N=N), 586(C-Br). 1H-NMR: 2.21(s, 3H, CH3), 5(s, 2H, N-CH2), 5.48(s, 2H, benzylic), 7.25-8.33(m, 13H, Ar-H and SO2-NH). 13C-NMR (δ ppm): 28(CH3), 33(NH-CH2), 52(C-benzylic),121-136(C-Ar), 158(C=O).
o-[N-(1-benzyl)-1,2,3-triazol-4-yl]methylsulfamoyl-N-(m-nitrophenyl) benzamide (9g)
The product 70%, m.p 180-181oC, IR cm-1: 3419(N-H), 1731(C=O), 1595(N=N), (1535) asym. (1336) sym. NO2.
o-[N-(1-(p-bromobenzyl))-1,2,3-triazol-4-yl]methylsulfamoyl-N-(m-nitrophenyl) benzamide (9h)
The product 72%, m.p 150-151 oC, IR cm-1: 3390(N-H), 1722 (C=O), 1595(N=N), (1539) asym. (1325) sym. NO2. 1H-NMR:4.99(s, 2H, N-CH2), 5.58(s, 2H,benzylic), 7.25-8.33(m, 13H, Ar-H and SO2-NH). 13C-NMR (δ ppm): 33(NH-CH2), 52(C-benzylic), 121-136 (C-Ar), 158(C=O).
o-[N-(1-benzyl)-1,2,3-triazol-4-yl]methylsulfamoyl-N-(o-nitrophenyl) benzamide (9i)
The product 70%, m.p 188-189oC, IR cm-1: 3440(N-H), 1726(C=O), 1627(N=N), (1460) (1330) NO2 (asym.) (sym.). 1H-NMR: 4.99(s, 2H, N-CH2). 5.58(s, 2H, benzylic) , 7.25-8.33(m, 13H, Ar-H and SO2-NH). 13C-NMR (δ ppm): 33(NH-CH2), 52(C-benzylic), 121-136 (C-Ar), 158(C=O).
o-[N-(1-(p-bromobenzyl))-1,2,3-triazol-4-yl]methylsulfamoyl-N-(o-nitrophenyl) benzamide (9j)
The product 68%, m.p 158-159oC, IR cm-1: 3407(N-H), 1722(C=O), 1593(N=N), (1560) (1325) NO2 (asym.) (sym.).
In vitro Cell viability Assay (MTT)32
The cytoxicity profiles were estimated by using “3-[4,5-dimethyl-thiazol-2-yl]- 2,5-diphenyl-tetrazolium bromide” (MTT) microculture, tetrazolium viability-method. To prepare stock solution we dissolve the compounds in DMSO and sequential dilution (12.5 μg/ml–200 μg/ml) were prepared through dissolving stock-solutions in cualture-cellular modelas (MCF7 cell line) breast-cancer. At the exact time in which after compounds (4a, 6a and 8f) treatment, MTT (5 mg/mL) was added in each wells and incubation of plate are 4 h. After removing the media, DMSO is add into each well for solubilized formazan-crystals. The same method is returned but using WRL-68 normal cell (liver cell) as negative control. Absorbance was measured at wavelength (575 nm) using (Hidex Chamealon platereader). Perceantage of cellular-viabilities were calculated with suitable control take into account. The concentration that inhibition 50% of cell-growth (IC50 value) is determined. All-tests are executed in triplicate. Percentage viability of cells exposed to various treatments was obtained as follows: cell Viability %=(Absorbance treated sample/ Absorbance of non-treated sample) ×100 (Non-treated cultures in all experiments contained the medium only).
Results and Discussion
TThe synthesized of Oxadiazole, Aza-β-lactam and 1,2,3-triazole derivatives were achieved by starting from compound (1a) that react with absolute ethanol, in the presence concentrated H2SO4 gave compound (2a) that have been reacted with 80% hydrazine hydrate to give o-(N-propargyl) sulfonamido benzohydrazide (3a). The absence of (C=O ester) stretching-band at (1737) cm-1 and the presence of new stretching-bands, at (3242-3330) cm-1and 1656 cm-1 that due to (NH-NH2) and (C=O amide) respectively are attributed to the formation of benzohydrazide derivative (3a), while 1H-NMR spectrum fig. (1) showed absence of signals 1.4ppm(CH3), 4.55ppm(O-CH2), and appearance of signals at 1.45ppm (NH2), 5.94ppm (NH-C=O) ppm. Cyclization of (3a) with (CS2) in presence (KOH) give 1,3,4-oxadiazole compound (4a) (scheme 2). Absence of stretching bands at (3242-3330) cm-1 for (NH-NH2) and (1656) cm-1 for (C=O amide) and presence weak stretching band of SH (2650) cm-1 and stretching band (C=N) (1596) cm-1 is attributed to the formation compound (4a), while 1H-NMR of compound (4a) fig. (2) display the following characteristic signals δ(ppm): 2.82(s, 1H, CH acetylinic), 3.8(s, 2H, CH2), 7.8-8.03(m, 4H, Ar-H), 8.2(s, 1H, NH-SO2).
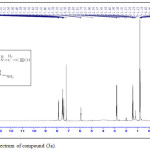 |
Figure 1: 1H-NMR spectrum of compound (3a). |
 |
Figure 2: 1H-NMR spectrum of compound (4a).
|
The reaction of compound (3a) with phenyl isocyanate to give compound (5a). The product was identified by FT.IR spectrum which shows absence of stretching band of (NH-NH2) at (3242- 3330) cm-1 and existence of new absorption bands due to (NH) at (3220) cm-1 and (C=O) at (1672) cm-1. Schiff-bases (6a-b) were synthesized by condensation of hydrazine derivative (3a) with various aromaticaldehydes (2,4-dimethoxybenzaldehyde and p-hydroxybenzaldehyde) with few drops of (glacial-acetic acid). The absence of (NH2) stretching bands at (3242-3330) cm-1 and appearance (C=N) stretching band at (1596-1602) cm-1 indicate the formation of Schiff-bases, while 1H-NMR spectrum showed singlet signal for (CH=N) at 9.1ppm. Moreover, The cyclization of (6a-b) with phenyl isocyanate via [2+2] cycloaddition reaction gave the corresponding “1,3-diazetidine-2-one (Aza-β-lactam)” (7a-b) derivatives. The disappearance of stretching band of (CH=N-) at (1614-1625) cm-1 and appearance stretching band of (C=O aza-β-lactam) at (1708-1712) cm-1 were utilized to confirm the compounds. Benzamide derivatives (8a-c) were synthesized by condensation compound (2a) with aniline derivatives such as (p-toluidine, o-nitroaniline and m-nitroaniline) in presence POCl3. The absence of absorption band of (O-H) at (3309) cm-1, and appearance absorp. band of (C=O amide) at (1735-1737) cm-1 gave good evidence for formation. While 1H-NMR spectrum of compound 8a fig. (3) showed appearance of signals at 2.85ppm (CH acetylinic), 6.5ppm (NH-C=O), the 13C-NMR of compound 8a fig. (4) display 27.46 (CH3), 75-76 (C C), 121-136 (C-Ar), 158 (C=O).
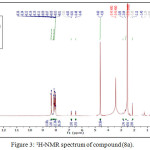 |
Figure 3: 1H-NMR spectrum of compound (8a). |
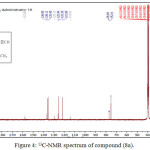 |
Figure 4: 13C-NMR spectrum of compound (8a). |
1,4-disubsituted-1,2,3-triazole derivatives (9a-j) (scheme 3) were prepared under click conditions by refluxing compounds 4a, 5a and (8a-c) with (benzyl and p-bromobenzyl) azide. Disappearance of absorption bands at (3274-3298) cm-1 for acetylenic C-H, (2125-2127) cm-1 for C C and 2096 cm-1 for azide group and appearance of new band for (N=N) at (1626-1562). While 1H-NMR and 13C-NMR spectra of compounds (9a), (9f), (9h) and (9i) exhibit the absence of singlet signal at (2.8) ppm due to (C-H) acetylenic fig. (5),(6),(8),(10) and disappearance of the signal at (75-76) ppm due to (C C) fig. (7),(9),(11) is good evidence for the formation of the 1,2,3-triazole derivatives.
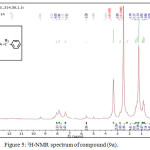 |
Figure 5: 1H-NMR spectrum of compound (9a). |
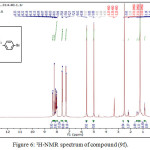 |
Figure 6: 1H-NMR spectrum of compound (9f). |
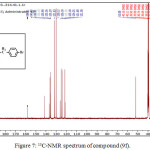 |
Figure 7: 13C-NMR spectrum of compound (9f). Click here to View figure |
 |
Figure 8: 1H-NMR spectrum of compound (9h). Click here to View figure |
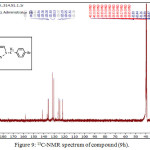 |
Figure 9: 13C-NMR spectrum of compound (9h). |
 |
Figure 10: 1H-NMR spectrum of compound (9i). Click here to View figure |
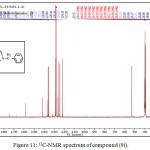 |
Figure 11: 13C-NMR spectrum of compound (9i). |
In-Vitro Cytotoxic Activity
The results of cytotoxic activity of compounds (4a, 6a and 9j) on MCF7 cancer cell were represented in Table 1 and Figures (12, 13 and 14) compared to normal cell line WRL-68 (liver cell) as negative control. The results showed that when we treated (MCF7) cell with different concentrations (12.5-200μg/ml) the best inhibition was at 100μg/ml by compound (12f) exhibited 71.66% of cancer cell death, with IC50 value 56.19μg/ml while exhibited 9.61% of normal cell death. As we comparison with another studying of anticancer potential [33] we find the best inhibition exhibited 51.4% at 100μg/ml on MCF7 cell line compared to 25 μg/ml doxorubicin as positive control so they didn’t use normal cells.
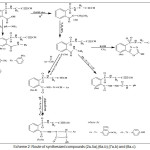 |
Scheme 2: Route of synthesized compounds (2a-5a),(6a-b),(7a-b) and (8a-c). |
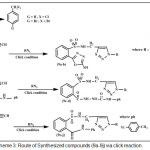 |
Scheme 3: Route of Synthesized compounds (9a-9j) via click reaction. |
Table 1: The anticancer activity of compounds 4a, 6a and 9j on MCF7 cell line by MTT method.
|
Concentrations of comp. (µg/ml) (Mean ± SD) |
||||||||||
|
Cell lines |
MCF-7 |
WRL-68 |
||||||||
|
Comp. no. |
200 |
100 |
50 |
25 |
12.5 |
200 |
100 |
50 |
25 |
12.5 |
|
4a |
66.09±8.21 |
40.81 ±7.73 |
6.58 ±6.45 |
0.16 ±4.85 |
2.69 ±2.74 |
35.77 ±7.37 |
7.51 ±4.45 |
1.19 ±3.03 |
2.57 ±0.89 |
0.69 ±2.49 |
|
6a |
67.01±1.67 |
52.49 ±9.80 |
15.36 ±7.59 |
_ |
_ |
41.83 ±12.46 |
_ |
1.51 ±2.68 |
0.42 ±4.02 |
0.67 ±4.75 |
|
9j |
84± 8.21 |
71.66 ±4.79 |
52.47±5.14 |
14.7± 3.57 |
3.35± 4.18 |
44.33 ±23.82 |
9.61 ±39 |
8.37 ±7.29 |
0.74 ±6.65 |
2.46 ±18.36 |
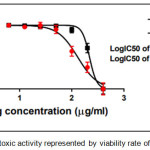 |
Figure 12: The cytotoxic activity represented by viability rate of comp. (4a) |
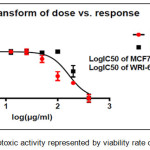 |
Figure 13: The cytotoxic activity represented by viability rate of comp. (6a). |
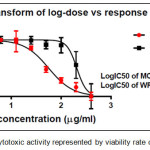 |
Figure 14: The cytotoxic activity represented by viability rate of comp. (9j). |
Conclusion
Some new oxadiazole, Aza-β-lactam derivatives were synthesized, and to obtained “1,4-disubstituted-1,2,3-triazole through click-chemistry”. The studying of cytotoxicity activity on some final prepared compounds (4a, 6a and 9j) showed that tested compounds have a promising significant (p≤0.05) cytotoxic activity. Compound 9j showed the best cytotoxic activity that has strong cell-growth inhibition with IC50 56.19μg/ml.
Acknowledgments
As the authors of this work, we would love to express our ample thankfulness and gratitude to the Department of Chemistry, College of Science, University of Baghdad and College of Pharmacy, for their magnificent support and providing us with facilities to conduct this research on all accounts.
References
- Raut, A.B.; Matto, M. R.; Acta. Chim. Solv., 2008, 55,448.
- Babak, K.; Leila, M.; Tetrahedron lett., 2011, 52,6424-6426.
CrossRef - Noha, I.Z.; Fabio, S .; Stefamo, F.; European. J. Med. Chem., 2010, 45,4523.
CrossRef - Dali P.; Gautum, P.; Angpla, K.C.; European. J. Med. Chem., 2011,46, 3085-3092.
CrossRef - Gopal, L.K.; Jasmine, K.; Varnm, K.; Bio. Organic. Med. Chem let., 2012, 22,1912-1916.
- John, K.A.; Veeramani, V.P.; Sharmila, N. S.; Tetrahedron lett, 2009, 65, 9989-9996.
CrossRef - Aoyama, Y.; Uenaka, M.; Konoike, T.; Nakayima, M.; Bioorganic and Medicinal chemistry letters, 2001, 11, 1691.
CrossRef - James, A.H.; Glenn, S.T.; Robert, T.; Roger, M.E.; J. Antimicr. Chemoth. , 2007, 4, 923.
- Srivastava, S.K.; Srivastava, S.L.; Srivastava, S.D.; Indian J. Chem., 2000, 39, 464.
- Vaishnav, P.; Demain, A.; Biotechnol. Adv., 2010, 7, 50.
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B.; Angew. Chem. Int. Ed., 2001, 40, 2004- 2021.
CrossRef - Kolb, H. C.; Sharpless, K. B.; Drug Discov. Today, 2003, 8, 1128-1137.
CrossRef - Moses, J. E.; Moorhouse, A. D.; Chem. Soc. Rev., 2007, 36, 1249-1262.
CrossRef - Tornøe, C. W.; Christensen, C.; J. Org. Chem., 2002, 67, 3057-3064.
CrossRef - Rostovtsev, V. V.; Green, L. G.; Fokin, V.V.; Sharpless, K.B.; Angew. Chem. Int. Ed., 2002, 41, 2596-2599.
CrossRef - Hiemstra, V. D.; Maarseveen, H. van; Eur. J. Org. Chem., 2006, 51-68.
- Wu, P.; Fokin, V. V.; Aldrichim. Acta., 2007, 40, 7-17.
- Meldal, M.; Tornøe, C. W.; Chem. Rev., 2008, 108, 2952-3015.
CrossRef - Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn M.G.; J. Am. Chem. Soc., 2003,125, 3192.
CrossRef - Manetsch, R.; Krasinski, A.; Radic, Z.; Raushel, J.; Tylor, P.; Sharpless, K.B.; Kolb H.C.; J. Am. Chem. Soc., 2004,126,12809.
CrossRef - Helms, B.; Mynar, J. L.; Hawker, C. J.; Fréchet, J.M.; J. Am. Chem. Soc., 2004, 126, 15020.
CrossRef - Karim, A. M.; Park, J. S.; DSSCs. Macromol Chem Phys., 2008, 209, 1967.
CrossRef - Schirrmacher, R.; Wangler, C.; Schirrmacher, E.; Mini-Rev Org Chem., 2007, 4, 317.
CrossRef
- Hussein, F.; Hello, K. M.; J. of Chem., 2002, 26(1), 35-41.
- Abdulla, A. F.; MSc. Thesis, University of Baghdad, College of Science, 2015.
- Hasan, A.; Thomas, N. F.; Gapil, S.; Molecules, 2011, 16(2), 1297-1309.
CrossRef - El– Tamaty, E. S.; Abdel– Fattah, M. E. et al.; Indian J. Chem., 1996, 35, 1067.
- Naser, A. W.; Iraqi National Journal of Chemistry, 2013, 50, 199.
- Furniss, B.; Hannaford, A. H.; Smith, P.; Tatchell, A.; Vogel’s Text book of practical organic chemistry, 5th Ed., Addison Wesley Longman, 1998, 1077.
- Issac, Y. A.; Mohamed, S. K.; Eissa, A. M.; Tantawya, A. H.; EL-Sawya, A. A.; Journal of Chemical and Pharmaceutical Research, 2012, 4(5), 2744-2750.
- Shneshil, M. K.; Ph.D. Thesis, University of Babylon, College of Science, 2016.
- Denizot, F.; Lang, R.; J. of Immunological Methods, 1996, 89(2), 271–277.
CrossRef - Alshanon, A. F. ; Firas, H.; Abdul hameed, A. and Alsaffar, A. Z.; International Journal of Pharma Sciences, 2015, 5(1), 904-910.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


