Enhancing the Light Harvesting Efficiency, open Circuit Voltage and Stability of Molybdenum Doped (Zno)6 Nanocluster in Dye-Sensitized Solar Cells: A DFT Study
S. Dheivamalar1 and K. Bansura banu1,2
and K. Bansura banu1,2
1Department of Physics, Periyar E.V.R. College (Autonomous), Tiruchirappalli-620023, India.
2Department of Physics, Holy Cross College (Autonomous), Tiruchirappalli-620002, India.
Corresponding Author E-mail: sitrulinsiragugal@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/340509
Article Received on : 07-08-2018
Article Accepted on : 04-10-2018
Article Published : 18 Oct 2018
In this study, the electronic and structural properties of drum structured Mo-doped Zn6O6 (MoZn5O6) cluster as the π conjugated bridging in the dye-sensitized solar cells (DSSC) were compared with its pristine form by density functional theory (DFT) calculations under Gaussian 09 Program. The frontier molecular orbital study was explored to determine the charge transport characteristics of donor-acceptor moieties over the entire visible range and the electron injection from the valence band (LUMO) orbital to the conduction band (HOMO) orbital of MoZn5O6. The energy gap (Eg), binding energy (EB), global reactivity descriptors, thermodynamic parameters and the dipole moment were also calculated for MoZn5O6 and compared with Zn6O6. The density of states (DOS) of MoZn5O6 material was investigated to demonstrate the importance of d orbital of Mo atom in hybridization. To examine the charge distribution, Mulliken atomic charge distribution and molecular electrostatic potential (MEP) were analyzed. A spectroscopic study was included for the better perception of the interaction of Mo with Zn6O6 cluster. The increased value of the first-order hyperpolarizability of MoZn5O6 from its pure clustermanifests the MoZn5O6 is a better candidate with the superior nonlinear optical property. The analysis of UV-Vis spectra through the time-dependent density functional theory (TD-DFT) discovers that the MoZn5O6 has larger light harvesting efficiency (LHE) which influences the higher photon to current conversion efficiency. As a result, the valence band (LUMO) of MoZn5O6 is intense than the conduction band (HOMO) of MoZn5O6 making an increase in the open circuit voltage (VOC) and hence it confirms that the MoZn5O6 material can be a used in photovoltaic applications.
KEYWORDS:MoZn5O6; Nanocluster; Photovoltaic applications; Thermodynamic parameters; Zn6O6
Download this article as:| Copy the following to cite this article: Dheivamalar S, Banu K. B. Enhancing the Light Harvesting Efficiency, open Circuit Voltage and Stability of Molybdenum Doped (Zno)6 Nanocluster in Dye-Sensitized Solar Cells: A DFT Study. Orient J Chem 2018;34(5). |
| Copy the following to cite this URL: Dheivamalar S, Banu K. B. Enhancing the Light Harvesting Efficiency, open Circuit Voltage and Stability of Molybdenum Doped (Zno)6 Nanocluster in Dye-Sensitized Solar Cells: A DFT Study. Orient J Chem 2018;34(5). Available from: http://www.orientjchem.org/?p=51048 |
Introduction
Nanostructured materials are currently of prodigious attention due to their reduced dimensions and are majorly take part in a large number of new devices. Among the renewable energy sources, solar energy is the favorable and sustainable alternative. Several photovoltaic devices are manufactured on II-VI semiconductor nanostructures.1,2 Dye-sensitized solar cells (DSSC) are the real replacement to the well established silicon-based solar cells owing to their greater incident photon to current conversion efficiency, low cost, ease of device fabrication and durable stability. The DSSCs are hinged on the absorption of photon energy over wide ranges and dye sensitizers. The π conjugated elements are used to stimulate the photosynthesis because of their electrochemical properties.3 Recently, the evolution of ZnO nanomaterials are growing exponentially, which was proven by the ZnO related papers in the literature.4,5 The ZnO is a semiconductor metal oxide with broad energy gap has a higher probability in field effect transistors, solar cells, gas sensors, photodetectors, photocatalysts and optoelectronics due to its higher excitation binding energy.6,7 The ZnO nanomaterials have replaced the bulk materials through their nanomorphology, functionality, low toxicity, good thermal stability, oxidation resistivity, biocompatibility, larger surface to volume ratio, higher chemical reactivity, and greater electron mobility.8,9 To extend the range of uses of ZnO nanomaterial, there are novel procedures to rephrase the electronic structures of ZnO such as depositing or doping various atoms in bulk ZnO materials experimentally and theoretically.10
The introduction of Mo atom into the ZnO material is an elemental reach to refine the properties of the host material.11 Doping mechanism is the introduction of substitutional impurity by replacing single Zn atom by single Mo atom or single O atom by single Mo atom of the Zn6O6 at any position causes the significant upgrade in optoelectronics successfully.12,13 The n-type Zn6O6 semiconductor material doping with an element of group VI as Mo, its physical properties can be tuned finely. Mo plays an essential character in the modern semiconductor industry and it is a predominant doping material to upgrade the performance of ZnO nanostructured materials. Recently, Chang-Lin Yu et.al.,14 has prepared Mo-doped ZnO photocatalysts by a grinding calcination method. They analyzed that, the Mo doping provided higher photocatalytic activity and stability than pure ZnO nanoparticles. R. Swapna et. al.,15 and V. Gokula Krishnan et. al.,16 have studied Mo-doped ZnO thin films by using the spray pyrolysis technique. They showed that the films have shown minimum resistivity, maximum carrier concentration and high electrical conductivity upon Mo doping.
In the present work, the Mo-doped Zn6O6 nano-cluster was studied and compared with its pristine form through energy, geometry, binding site, electronic properties, HOMO/LUMO energies, Mulliken charge distribution, DOS spectra, simulated IR, UV-Vis spectra and molecular electrostatic potential. A challenging way to inscribe this study is to utilize DFT and TD-DFT computational methods.17 The achievement of the solar cells was strengthened by the π conjugated system of the bridge. The structural unit of dyes is represented as D-π-A system. The Zn6O6 interacts with Mo which intensifies the light harvesting efficiency, open circuit voltage, the electron injection and the stability of solar cells. The MoZn5O6 material discovers extensive requirements in solar cells since an efficient photosensitizer which harvest the sunlight and retard the charge combination.
Computational Methods
Geometrical optimization of pure Zn6O6, Single Mo atom and MoZn5O6 were analyzed using density functional theory (DFT) method. The energy of HOMO (EHOMO), the energy of LUMO (ELUMO), and the energy gap (Eg = ELUMO – EHOMO) were investigated for pure Zn6O6 and MoZn5O6 using the hybrid density functional B3LYP and correlation functional of Lee- Yang- Parr with 6-31G and LANL2DZ basis sets using Gaussian 09 program.18-20 For pure Zn6O6 and MoZn5O6 material, electronic states for a low spin as the singlet state (spin multiplicity = 1) were applied. To evaluate the excitation energy, UV-Vis electronic transition, light harvesting efficiency and oscillator strength of photosensitizers, TD-DFT calculations were employed at the CAM-B3LYP/6-31G level. The density of states (DOS) for pure Zn6O6 and MoZn6O6 nanostructures were plotted by employing Gauss Sum Program.21 The determination of dipole moment, chemical parameters and polarizability were employed by the B3LYP/6-31G in Gaussian 09 program.22 In further, natural bond orbital (NBO) analysis, molecular electrostatic potential (MEP) analysis and Mulliken charge distribution of each atom in the title compounds were also implemented in the Gaussian 09 program. The thermodynamic parameters and the vibrational frequency were computed at the same level of theory.23 The vibrational frequencies were evaluated to get all the structures are at a local minimum. Excited states analysis were performed using Time Dependent-Density Functional Theory (TD-DFT) Kohn-Sham equations in Gaussian 09 program.24 The interaction between Mo and Zn6O6 cluster can be achieved by calculating binding energy as
EB = EMo-Zn6O6 – EMo – EZn6O6 (1)
Where EMo-Zn6O6 is the total energy of MoZn5O6 cluster, EMo is the total energy of the isolated Mo atom and EZn6O6 is the total energy of the isolated Zn6O6 cluster.25 The negative value of the binding energy stipulates the exothermic process.26
The energy gap Eg is defined as,
Eg = ELUMO – EHOMO (2)
Where, EHOMO and ELUMO are the energies of HOMO and LUMO, respectively. The energy shift ∆Eg was calculated as the ratio of the difference of Eg1 measured in pure Zn6O6 (reference value) and Eg2 measured in MoZn5O6 with respect to the reference value.27
ΔEg = [(Eg2 – Eg1) / (Eg1)] (3)
The middle of the energy gap Eg is the chemical potential (µ). The chemical potential (µ) of free gas of electron is equal to its Fermi level. The Fermi level of MoZn5O6 was determined as the center of the energy gap Eg.28
Results and Discussion
Structures and Stability
The optimized geometries of four isomeric structures of pure Zn6O6 nanocluster were obtained by using the Gaussian 09 program in the singlet ground state are shown in Fig. 1, whereas Fig. 2 represents the optimized drum structured Zn6O6 with MoZn5O6. The Zn6O6 nanocluster has four structural variants. Among all the structures, the properties of three-dimensional drum structured Zn6O6 have been scrutinized in this manuscript. The most known ZnO presentations are related to hexagonal forms and we expect that Zn6O6 nanocluster represents a superior potential source for further more research.29 The structural data in the ground state and excited state of pure Zn6O6 and MoZn5O6 were listed in Table 1.
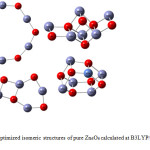 |
Figure 1: The optimized isomeric structures of pure Zn6O6 calculated at B3LYP/6-31G level of theory. |
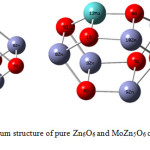 |
Figure 2: The optimized drum structure of pure Zn6O6 and MoZn5O6 calculated at B3LYP/6-31G level of theory. |
Table 1: The calculated bond length of Zn6O6 and MoZn5O6 at B3LYP/6-31G in the ground state (Gnd) and excited state (Exc) analysis.
| Bond distance | Zn6O6 (grd) | Zn6O6 (Exc) | Bond distance | MoZn5O6 at Zn(12) (grd) | MoZn5O6 at Zn(12)(Exc) |
| Zn(1)-O(2) |
1.93 |
1.91 |
Zn(1)-O(2) |
1.89 |
1.9 |
| Zn(1)-O(7) |
1.99 |
1.93 |
Zn(1)-O(7) |
1.94 |
1.96 |
| Zn(1)-O(9) |
1.99 |
1.93 |
Zn(1)-O(9) |
1.94 |
1.96 |
| O(2)-Zn(10) |
1.99 |
1.93 |
O(2)-Zn(10) |
1.94 |
1.96 |
| O(2)-Zn(12) |
1.99 |
1.93 |
O(2)-Mo(12) |
1.74 |
1.77 |
| Zn(3)-O(4) |
1.93 |
1.91 |
Zn(3)-O(4) |
1.88 |
1.9 |
| Zn(3)-O(8) |
1.99 |
1.93 |
Zn(3)-O(8) |
1.95 |
1.96 |
| Zn(3)-O(9) |
1.98 |
193 |
Zn(3)-O(9) |
1.95 |
1.97 |
| O(4)-Zn(10) |
1.99 |
1.93 |
O(4)-Zn(10) |
1.94 |
1.96 |
| O(4)-Zn(11) |
1.99 |
1.94 |
O(4)-Zn(11) |
1.95 |
1.96 |
| Zn(5)-O(6) |
1.93 |
1.91 |
Zn(5)-O(6) |
1.89 |
1.91 |
| Zn(5)-O(7) |
1.99 |
1.94 |
Zn(5)-O(7) |
1.95 |
1.97 |
| Zn(5)-O(8) |
1.99 |
1.93 |
Zn(5)-O(8) |
1.95 |
1.97 |
| O(6)-Zn(11) |
1.98 |
1.93 |
O(6)-Zn(11) |
1.95 |
1.96 |
| O(6)-Zn(12) |
1.98 |
1.93 |
O(6)-Mo(12) |
1.74 |
1.74 |
| Zn(10)-O(9) |
1.93 |
1.91 |
Zn(10)-O(9) |
1.89 |
1.91 |
| Zn(11)-O(8) |
1.93 |
1.91 |
Zn(11)-O(8) |
1.88 |
1.91 |
| Zn(12)-O(7) |
1.93 |
1.91 |
Mo(12)-O(7) |
1.74 |
1.74 |
The nanostructure of drum structured pure Zn6O6 cluster has two hexagonal rings and six tetragons in the singlet ground state.30 The symmetry of the Zn6O6 cluster is C1 point group symmetry. Two different kinds of Zn-O bond lengths were identified within the pure Zn6O6. The bond distance of 1.93 Å was mutually shared between the two hexagons and the bond length of 1.99 Å was observed between the tetragon and a hexagon in DFT optimization. This bond length prediction is fine accordance with the Zn-O bond was calculated by Birajdar et. al.,31 and seetawan et.al.,32 depending on the structures of nanocluster. Results of TD-DFT the Zn-O bond length for pure Zn6O6 cluster between 1.91Å-1.93Å is fine agreement with the theoretical studies by wang et.al.33 The charge transfer from the Zn atom to the O atom in pure Zn6O6 results the ionic bond of Zn-O. The bond angles of tetragons and the hexagons in the pure Zn6O6 cluster varied from 89º to 910 and 116º to 1240. The calculated values of vibrational frequencies are positive from70cm-1 to 716 cm-1.
Analyzing the optimized structure of MoZn5O6, the bond distances of Zn-O bonds are increased in size when Mo replaced oxygen atoms at any position. The replacement of Zn by Mo at Zn (12) position favors the stabilization of the material by reducing bond distances in the singlet ground state is comparable to the work by Woodley et. al.34 They showed Mg and Cd doping with pure ZnO nanoclusters to study their stability. The larger bond length difference occurs among the two hexagons at Zn(1)-O(7), Zn(10)-O(2), Zn(10)-O(4), Zn(5)-O(8) bonds to 1.88Å and the bond distance between the tetragon and hexagon of Zn(11)-O(8) reduced to 1.94Å. The bond distance of Mo(12)-O(7), Mo(12)-O(2), Mo(12)-O(7) was calculated as 1.74Å. Results of TD-DFT for Mo doping at Zn(12) position makes the changes in bond length at Mo(12)-O(2) increases to 1.77Å and Zn(5)-O(8), Zn(10)-O(2) to 1.96Å. The replacement of Zinc by Mo at Zn(10) brings the changes in bond distance among the hexagons as 1.91Å at Zn(1)-O(2), Zn(5)-O(6), Zn(10)-O(9) and the bond distance between the tetragon and a hexagon to 1.96Å at O(2)-Zn(10), Zn(5)-O(8) in the singlet DFT calculation. TD-DFT calculation brings the changes in Zn(10)-O(9) to 1.90Å and Zn(10)-O(2) to 1.95Å while Mo doping at Zn(10) position. The replacement of Zinc by Mo at Zn(3) and Zn(11) position elongates the bond distance among the two hexagons as 1.96Å and the bond distance connecting the hexagon and the tetragon as 2.01Å in the ground state and decreases at excited state analysis. The doping of Mo at Zn(5) and Zn(1) position produces a slight variation in the bond length than the pure cluster in both DFT and TD-DFT calculations. The huge difference in the bond length of Mo substituted structure and the pure form was observed at Zn (12) position of Mo doping in the singlet ground state. The interaction between Mo and the Zn6O6 nanocluster were studied by calculating the corresponding binding energy EB as -1.76 Kcal/Mol. This calculation reveals that there were exothermic interaction with negative binding energy.35
Frontier molecular orbital theory
The π type molecular orbital characteristics were exhibited by the HOMO/LUMO contours. The bonding behavior was specified by the HOMO orbital and the anti-bonding features were enumerated by the LUMO orbital. The HOMO/LUMO transition is identified as π – π* intramolecular interaction.36 Fig. 3 shows the distribution of HOMO and LUMO of pure Zn6O6 and MoZn5O6. The Zn atoms in the cluster have a positive charge which is an acceptor and O atoms have a negative charge which is the donor. The positive region appeared in green color and the negative region appeared in red color. The energies of HOMO and LUMO for pure Zn6O6 were -6.16eV and -2.89eV with energy gap Eg of 3.27eV was inadequate concurrence with the study done by Anitha et. al.,37 The high energy gap of pure cluster indicates the thermal stability and inertness towards reactivity. It specifies that the electron in the valence band requires more energy to go to the conduction band. The larger energy gap makes the ZnO nanocluster applied in electronic devices.
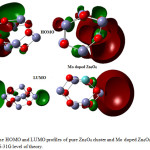 |
Figure 3: The HOMO and LUMO profiles of pure Zn6O6 cluster and Mo doped Zn6O6 calculated at B3LYP/6-31G level of theory. |
After substitution with Mo, the HOMO levels are mostly scattered along the Mo doping region. The HOMO and LUMO describe the charge transfer between Mo and Zn6O6 cluster. The energies of HOMO and LUMO are changed to -4.64eV, -0.475eV and the energy gap is 4.16eV. 38 The HOMO of the MoZn5O6 significantly shifted to the higher energies by -1.52eV. The photovoltaic properties of the above compound in solar cell applications were analyzed because of the extraordinary property of ZnO nanocluster as a shallow donor or acceptor.39 The LUMO energy of the MoZn5O6 was higher than the conduction band (HOMO) of MoZn5O6, which predicts that this compound may be an effective candidate in a photovoltaic cell uses. The open circuit voltage of the organic solar cells is linearly related with the HOMO of the donor and LUMO of the acceptor.40 The open circuit voltage VOC was calculated by,
VOC = │EHOMO (Donor)│–│ELUMO (Acceptor)│ -0.3 (4)
The dye MoZn5O6 takes up the photon of energy which drives electrons to LUMO of the dye and then to the conduction band of MoZn5O6. The VOC of pure Zn5O6 and MoZn5O6 were calculated to be 3.57eV and 4.46eV. The MoZn5O6 has high VOC and the MoZn5O6 was used as efficient photosensitizers due to the good light absorption characteristics. All the methods of calculation used here are affirming that the energy gap of MoZn5O6 material is sensitive towards Mo doping. The energy gap values of Zn6O6 and MoZn5O6 were calculated at B3LYP/6-31G, B3LYP/6-311G, B3LYP/LANL2DZ has been listed in Table 2.
Table 2: The values of HOMO and LUMO energies (EHOMO and ELUMO), an energy gap (Eg) and the energy shift (∆Eg) of Zn6O6 and MoZn5O6 calculated at B3LYP/6-311G, 6-31G and the LANL2DZ level of theory.
| System | Basis set | EHOMO (eV) | EFeV | ELUMO (eV) | Energy Gap Eg (eV) | ∆EgeV |
| Zn6O6 | B3LYP/6-31G |
-6.16 |
-4.52 |
-2.89 |
3.27 |
– |
| MoZn5O6 |
-4.64 |
-2.79 |
-0.475 |
4.16 |
0.89 |
|
| Zn6O6 | B3LYP/6-311G |
-5.94 |
-4.42 |
-2.79 |
3.14 |
– |
| MoZn5O6 |
-4.65 |
-2.58 |
-0.521 |
4.12 |
0.98 |
|
| Zn6O6 | B3LYP/LANL2DZ |
-5.82 |
-4.27 |
-2.73 |
3.08 |
– |
| MoZn5O6 |
-4.89 |
-2.67 |
-0.469 |
4.42 |
1.34 |
Density of State Analysis
The density of states plot of pure Zn6O6 and MoZn5O6 are shown in Fig. 4. The energy gap of pure Zn6O6 was determined as 3.27eV. The energy gap MoZn5O6 was calculated as 4.16eV. By comparing the HOMO and LUMO levels of the pure Zn6O6 and MoZn5O6, the doping process causes the significant shift of HOMO of pure Zn6O6 to higher energy levels and the new HOMO of MoZn5O6 lies between the HOMO and LUMO of the pure Zn6O6. The DOS plot of MoZn5O6 indicates that Mo atom significantly participates in having a higher value of Eg and VOC compared to the pure cluster. The Fermi energy (EF) was increased after Mo doping and hence EF shifts towards the conduction band. The EF was increased from -4.52eV to -2.79eV after Mo doping. This increase in Fermi level assists the decrease in work function causes the MoZn5O6 material is an essential candidate in field emission. After the Mo substitution over the Zn6O6 nanocluster, Mo enhances the VOC of MoZn5O6 by increasing the energy gap compared to the pure Zn6O6 cluster. Therefore, the MoZn5O6 material was found remarkable applications in solar cell devices.
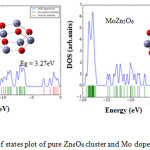 |
Figure 4: The density of states plot of pure Zn6O6 cluster and Mo doped Zn6O6 nano material. |
Charge Population Analysis
Mulliken atomic charge distribution affords the information about the electron population of atoms in Zn6O6 and MoZn5O6 cluster. The Mulliken charge values were calculated from B3LYP/6-31G basis set. The Mulliken atomic charges of individual atoms present in Zn6O6 and MoZn5O6 are given in Table 3. For a pure Zn6O6 cluster, Zn1, Zn3, Zn5, Zn10, Zn11, and Zn12 atoms possess positive charges which are acceptor atoms and the atoms O2, O4, O6, O7, O8, and O9 own negative charges which are donor atoms. The oxygen atom (O7) in Zn6O6 has a more negative charge which is a donor. The Zinc atom (Zn10) has a more positive charge which is an acceptor. For MoZn5O6, Zn1, Zn3, Zn5, Zn10, Zn11, and Mo12 atoms possess positive charges which are acceptor atoms and the atoms O2, O4, O6, O7, O8, and O9 hold negative charges which are donor atoms.41 The Zn1 atom manifests more positive charge with a value 0.973e and O7 atom manifests more negative charge with a value 0.972e. The Mulliken atomic charge distributions of pure Zn6O6 and MoZn5O6 have been presented in Fig. 5. The NBO charges of pure Zn6O6 and MoZn5O6 in the singlet ground state are also shown in Table 3. In MoZn5O6, the charge transfers from the dopant Mo to the remaining part of the material as compared with the atomic charge of Zn atom of undoped Zn6O6. The metal dopant atom Mo acts as an electron acceptor.
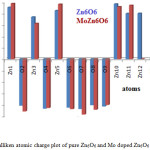 |
Figure 5: Mulliken atomic charge plot of pure Zn6O6 and Mo doped Zn6O6 nano cluster. |
Table 3: Mulliken atomic charges (Mul) and NBO charges of Zn6O6 and MoZn5O6 calculated at B3LYP/6-31G.
| Atom | Zn6O6 (Mul) | MoZn5O6 (Mul) | Zn6O6 (NBO) | MoZn5O6 (NBO) |
| Zn1 |
0.905 |
0.972 |
1.6 |
1.379 |
| O2 |
-0.817 |
-0.917 |
-1.419 |
-1.232 |
| Zn3 |
0.735 |
0.622 |
1.503 |
1.432 |
| O4 |
-0.869 |
-0.857 |
-1.515 |
-1.482 |
| Zn5 |
0.849 |
0.959 |
1.404 |
1.483 |
| O6 |
-0.851 |
-0.877 |
-1.524 |
-1.231 |
| O7 |
-0.875 |
-0.971 |
-1.475 |
-1.136 |
| O8 |
-0.808 |
-0.887 |
1.517 |
-1.438 |
| O9 |
-0.83 |
-0.805 |
-1.512 |
-1.272 |
| Zn10 |
0.964 |
0.915 |
1.469 |
1.457 |
| Zn11 |
0.797 |
0.94 |
1.593 |
1.414 |
| Zn12 |
0.8 |
0 |
1.392 |
0 |
| Mo |
0 |
0.905 |
0 |
0.625 |
Analysis of Chemical parameters
The ionization potential (I), electron affinity (A), chemical hardness (η), chemical softness (S), chemical potential (µ), and electrophilicity index (ω) of MoZn5O6 were determined and compared with the pure Zn6O6 cluster and their values are tabulated in Table 4.The global reactivity descriptors were estimated from the energies of HOMO and LUMO to study the chemical stability and reactivity of MoZn5O6.
Table 4: Global Reactivity Descriptors of pure and MoZn5O6 calculated at B3LYP/6-31G.
| Property | Zn6O6 | MoZn5O6 |
| I = -Eh eV |
6.16 |
4.64 |
| A = -El eV |
2.89 |
0.475 |
| Ƞ = (I – A)/2eV |
1.63 |
2.08 |
| µ = – (I + A)/2eV |
-4.52 |
-2.55 |
| Ψ = -µeV |
4.52 |
2.55 |
| S = 1/2ƞ eV |
0.3 |
0.24 |
| ω = µ2/2ƞeV |
6.26 |
1.56 |
| Dipole Moment |
0.18 |
1.86 |
| Polarizability |
-92.08 |
-108.63 |
| Hyper Polarizability |
3.17 |
62.74 |
On the basis of Koopman’s theorem,42 the energies of HOMO (EHOMO) and LUMO (ELUMO) are associated with the ionization potential (I) and the electron affinity (A) as I = -EHOMO and A = -ELUMO. The ionization potential reduces when a Mo atom was substituted on Zn6O6 nanocluster. The ionization potential of the pure Zn6O6 was 6.16eV which declined to 4.64eV for MoZn5O6. The electron affinity decreases from 2.89eV to 0.475eV after Mo doping. This calculation correlated with Phool Singh Yadav et. al.,43 as the ionization potential (I) was greater than the electron affinity (A). The chemical potential (µ) is defined based on the equation,
µ = – (I+A)/2 (5)
The chemical potential (µ) increases for MoZn5O6 than its pure cluster. The calculated value of chemical potential (µ) for pure Zn6O6 was -4.52eV and -2.55eV for MoZn5O6. The Fermi level (EF) of the MoZn5O6 is sited at the nucleus of the energy gap Eg. The chemical potential (µ) is also the middle point of the energy gap Eg, therefore Fermi level is similar to the chemical potential (µ). The electronegativity (Ψ) is the negative of the chemical potential (µ).
The chemical hardness (η) was calculated by,
η = (I – A)/2 (6)
The chemical hardness (η) of Zn6O6 cluster was calculated as 1.635eV. The higher value of chemical hardness (η) in Zn6O6 predicts its rigid chemical stability. The chemical hardness (η) increases to 2.08eV after Mo substitution over the cluster. The chemical hardness (η) depends on the ionization potential (I) and electron affinity (A). The chemical softness (S) and the electrophilicity index (ɷ) are explained as,
S = 1/2ƞ (7)
ω = µ2/2ƞeV (8)
The chemical softness (S) is a perceptual of the chemical hardness (η). The chemical softness (S) of Zn6O6 was calculated as0.30eV. The chemical softness (S) decreases to 0.24eV when a Mo atom was substituted on the pure Zn6O6 cluster. The electrophilicity index (ɷ) of Zn6O6 was calculated as 6.26 eV and 1.56eV for MoZn5O6. The electrophilicity index (ɷ) computes the tendency of the material to accept electrons.
Analysis of NLO Parameters
The dipole moment is a salient electronic property which explains the charge distribution of the system. The calculated dipole moment for the pure cluster was found as 0.189 Debye and 1.867 Debye for MoZn5O6. The high value of dipole moment predicts the strong interaction among Mo and Zn6O6 cluster and it favors the greater NLO property. The polarizability and the first order hyperpolarizability were determined using DFT calculations.44
The hyperpolarizability was expressed as,
µ = (µx2+µy2+µz2)1/2 (9)
The total hyperpolarizability,
βTOTAL = (βx2+βy2+βz2)1/2 (10)
The mean polarizability,
<α> = 1/3(αxx+αyy+αzz) (11)
The calculated mean polarizability of pure Zn6O6 was -92.0865 Debye. The total hyperpolarizability of Zn6O6 was found to be 3.1712 Debye. The calculated mean polarizability of MoZn5O6 was -108.6326 Debye. The calculated total hyperpolarizability of MoZn5O6 was 62.74Debye. The highest contribution of the αzz component indicates, the pure and Mo-doped Zn6O6 cluster elongated towards the Z direction and contracted to the X direction. The βxzz component contributes with a larger part of hyperpolarizability in the pure form with the value 9.6624. The βxxy component contributes more towards the hyperpolarizability of MoZn5O6 with the value 20.0258. The MoZn5O6 material is a captivating gadget for further research of nonlinear optical properties due to the doping of the Mo atom.
Thermodynamic parameters:
The thermodynamic parameters of pure Zn6O6 and MoZn5O6 are displayed in Table 5. The thermal capacity (CV) of MoZn5O6 was increased to 45.07 Cal/Mol-Kelvin from 43.86 Cal/Mol-Kelvin as compared to Zn6O6 cluster. The CV shows that, the thermal capacity of the Zn6O6 and MoZn5O6 at room temperature and constant volume. The value of zero-point vibrational energy of Zn6O6 and MoZn5O6 were found to be 16.77 and 15.94 Kcal/Mol. The change in enthalpy (ΔH), the change in entropy (ΔS) and the change in Gibbs free energy (ΔG) of pure Zn6O6 and MoZn5O6 were computed from the frequency calculations using the consequent equations,
ΔH = HMo-Zn6O6 – HMo – HZn6O6 (12)
ΔS = SMo-Zn6O6 – SMo – SZn6O6 (13)
ΔG= GMo-Zn6O6 – GMo – GZn6O6 (14)
Where, HMoZn5O6, HZn6O6, and HMo are the sum of electronic and thermal enthalpies of Zn6O6, MoZn5O6, and Mo atom. GMoZn5O6, G Zn6O6, and GMo are the sum of electronic and thermal free energies of Zn6O6, MoZn5O6, and Mo atom. S MoZn5O6, SZn6O6, and SMo are the entropies of Zn6O6, MoZn5O6, and Mo atom.45 The values of ∆H, ∆S and ∆G were calculated as 65.4454, 38.078 and 65.4239Cal/M-Kelvin.
Table 5: The thermodynamic parameters of pure Zn6O6 and MoZn5O6 calculated at B3LYP/ 6-31G.
| Thermodynamic Parameters | Zn6O6 | MoZn5O6 |
| Total energy (Thermal), Etotal (Kcal/mol |
24.56 |
23.68 |
| Vibrational energy, Evib(Kcal/mol) |
22.78 |
21.91 |
| Zero points vibrational energy (Kcal/mol) |
16.77 |
15.94 |
| Rotational constants (GHz) | ||
| X |
0.484 |
0.472 |
| Y |
0.342 |
0.305 |
| Z |
0.295 |
0.268 |
| Specific heat Cv (cal/mol/K) |
45.07 |
43.86 |
| Entropy S (cal/mol/K) |
112.41 |
113.99 |
| Zero point correction (Hartee/particle) |
0.026 |
0.025 |
| Thermal correction to energy |
0.039 |
0.038 |
| Thermal correction to the enthalpy |
0.04 |
0.038 |
| Thermal correction to Gibbs free energy |
-0.013 |
-0.015 |
Molecular electrostatic potential
Molecular electrostatic potential (MEP) predicts the three-dimensional charge distribution of Zn6O6 and MoZn5O6. All isosurfaces are depicted with isovalue of 0.0004e/au3 in Gauss view software.46 MEP plot offers the positive and the negative region of Zn6O6 cluster and MoZn5O6 are shown in Fig. 6. In Fig. 6a, the Zn atoms were positively charged which are present in blue color. The O atoms were negatively charged which are appeared in red color. This reveals that there is the charge transfer from Zn atoms to O atoms which results in ionic bonds in the Zn6O6 nanocluster. In Fig. 6b, based on the MEP calculation upon MoZn5O6 cluster, the Zn atoms were specified by red color shows negative in charge. This confirms that there is a charge transfer from Mo doping material to the nanocluster because of the strong interaction. The total charge of MoZn5O6 material was -1.0. MEP surface prompted by the charge distribution of Zn6O6 and MoZn5O6 at an atomic site is described as,
![]()
Where ZA is the charge on nucleus A located at RA .47
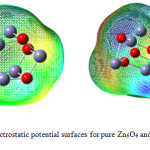 |
Figure 6: Molecular electrostatic potential surfaces for pure Zn6O6 and Mo substituted Zn6O6 nano cluster. |
Vibrational analysis
The vibration frequencies were the unique parameter to explore the local minimum in structures. The theoretical FTIR spectra of Zn6O6 and MoZn5O6 were presented in Fig. 7. Transmittance is the principal features of Zn-O vibration.48 There are 30 possible modes of vibrations in pure Zn6O6. The simulated IR spectrum of pure Zn6O6 shows the absorption peaks at 430cm-1, 451cm-1 and 547cm-1 have corresponded to the Zn-O stretching vibrations.49,50
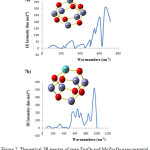 |
Figure 7: Theoretical IR spectra of pure Zn6O6 and MoZn5O6 nano material. |
Sowbagya et. al.,51 have synthesized Mo-doped ZnO nanoparticles and studied the FTIR spectrum. They showed that the Zn-O stretching vibration was observed at 443 cm-1. The existence of Mo-O3 depicted with the peak around 556cm-1, 859cm-1 and 617cm-1. The stretching vibration of Mo=O was observed at 925cm-1. In this present theoretical investigation, the Mo-doped Zn6O6 exhibits 30 modes of vibrations. The peak at 438 cm-1 was assigned to the stretching vibration of the Zn-O bond. The peaks at 533cm-1, 606cm-1 and 842cm-1 were assigned to the various bonds of Mo(12)-O(2), Mo(12)-O(6) and Mo(12)-O(7) respectively. The absorption band at 952cm-1 indicating the Mo=O stretching vibration. The presence of additional absorption bands from 438cm-1 to 1001cm-1 indicates the existence of Mo and its oxides.
Absorption Spectra and Photovoltaic properties:
The UV-Vis spectra of pure Zn6O6 and MoZn5O6 were analyzed and the wavelength (λmax), excitation energies (∆E) and oscillator strength (f) were computed using the singlet excited state at the B3LYP/6-31G by the time-dependent density functional theory calculations (TD-DFT) and the results were listed in Table 6.
Table 6: The excitation wavelength (λmax), excitation energies (∆E) and oscillator strength (f) of Zn6O6 and MoZn5O6.
| Compound | Excitations | Excitation wavelength (nm) | Oscillator strength (f) | Energy (eV) |
| Zn6O6 | 114 à115 |
578 |
0.6 |
2.14 |
| 113 à115 |
565 |
0.42 |
2.19 |
|
| 111 à115 |
531 |
0.15 |
2.33 |
|
| MoZn5O6 | 60à 62 |
511 |
0.12 |
2.43 |
| 60à 63 |
417 |
0.15 |
2.97 |
|
| 60à 62 |
412 |
0.9 |
3 |
The high absorption of the Zn6O6 and MoZn5O6 were studied from the energy gap (Eg). The lowest singlet to singlet excited state transitions were investigated to study the electronic absorption mechanism in whole visible/near-IR regions. On the basis of Frank Condon principle, the maximum peak correlates with vertical excitation. The electronic excitation observed as π – π* transitions. The energy gap of Zn6O6 cluster was 3.27 eV so that the λmax was fall in the visible region. There are three peaks with excitation energies 2.14eV, 2.19eV, and 2.33eV for the pure Zn6O6. The excitation wavelengths (λmax) of the absorption spectrum of Zn6O6 were determined as 578nm, 565nm, and 531 nm. The higher value of oscillator strength (f) was calculated as 0.60 with the excitation energy (∆E) of 2.14eV. After the Mo doping in Zn6O6 cluster, the excitation energies (∆E) were calculated as 2.43eV, 2.97eV, and 3eV. The higher value of oscillator strength (f) was calculated as 0.90 with the excitation energy 3eV. The MoZn5O6 materialis beneficent in increasing the light harvesting efficiency which leads to the higher short circuit current density (Jsc).52 The short-circuit current density (Jsc) is closely related to the charge conversion efficiency (η) of the solar cell can be calculated by the following equation,
Jsc = ∫LHE (λ) φInject.ηcollect dλ (16)
Where LHE (λ) is the light harvesting efficiency, φInject is the electron injection efficiency to the conduction band of MoZn5O6 and ηcollect is the electron collection efficiency. The φInject is associated with the driving force ΔGinject for the electron injection. The light harvesting efficiency LHE (λ) can be expressed as53
LHE = 1-10-f (17)
Where f is the oscillating strength of the maximum absorption spectra of Zn6O6 and MoZn5O6.
The large φInject leads to the higher Jsc. The φInject is directly proportional to the free energy of electron injection as
φInject α f (- ΔGInject)
ΔGInject = Edye* – ECB (18)
Where Edye* represent the oxidation potential energy in the excited state and ECB represent the reduction potential of the conduction band. Edye* can be estimated by,
Edye* = Edye – ΔE (19)
Where Edye represents the energy of oxidation potential in the ground state and ΔE is the energy of the electronic vertical transition corresponding to λmax. The oxidation potential of the dye is directly related to the HOMO level and the driving force for the reduction of oxidized dye increase at the larger oxidation potential. The values of an electron injection (ΔGInject), oxidation potential (Edye* and Edye) and open circuit voltage (VOC) of pure Zn6O6 andMoZn5O6 are summarized in Table 7.
Table 7: The values of an electron injection (ΔGInject), oxidation potential (Edye* and Edye) and open circuit voltage (VOC) of pure Zn6O6 andMoZn5O6 calculated from UV_VIS absorption spectra.
| System | Eg | VOC | Eoxdye(eV) | Eoxdye*(eV) | ∆GInject | LHE |
| Zn6O6 |
3.27 |
3.57 |
2.14 |
2.33 |
3.83 |
0.75 |
| MoZn5O6 |
4.16 |
4.46 |
2.43 |
3 |
1.64 |
0.87 |
Conclusion
The theoretical investigation on geometrical structures, binding nature, UV-Vis spectra, vibrational frequencies and the electronic properties of Zn6O6 andMoZn5O6 have been analyzed to predict the photovoltaic properties using DFT and TD-DFT theory. The doping of Mo makes the substantial modification in the HOMO and LUMO energies which ensures the open circuit voltage (VOC) of the MoZn5O6 was higher than the pristine Zn6O6 cluster. The high open circuit voltage making MoZn5O6 as a potential compound in the photovoltaic applications and is also been an efficient sensitizer. The density of state analysis brings the light absorption characteristics of MoZn5O6 system expands consequently by the doping material. The calculations from the various basis sets predict that there was a significant charge transfer from the dopant to the pure cluster, which enhances the energy gap Eg. The nonlinear optical characteristic of MoZn5O6 material was studied by hyperpolarizability. The dipole moment of MoZn5O6 was higher than its pure form. The increase in light harvesting efficiency of MoZn5O6 was calculated from the electronic transition in UV-Vis spectra. The replacement of Mo assists the hexagonal MoZn5O6 nanomaterial has more photoresponse than Zn6O6 which would be a better potential originator in solar cell applications.
References
- Gu, G. R., Ito, T., Field emission characteristics of thin metal coated nanosheet carbon films, Applied Surface Science., 2011, 257, 2455-2460.
CrossRef - Mackowski, S., Karczewski, G., Wojtowicz, Qju, X., Howe, J. Y., Mayer, H. M., Tuncer, E., Parantharaman, M. P., Thermal stability of HfO2 nanotube arrays, Applied Surface Science, 2011, 257, 4057-4081.
- Hagfeldt, A., Boschloo, G., Sun, L., Kloo, L., Pettersson, H., Dye-sensitized solar cells, Chem. Rev. 2010, 110 , 6595-6663.
CrossRef - Wu, Y. L., Tok, A. I. Y., Boey, F. Y. C., Zeng, X. T., Zhang, X. H., Surface modification of ZnO crystals, Applied Surface Science, 2007, 253, 5473-5479.
CrossRef - Prades, J. D., Cirera, A., Morante, J.R., Ab initio calculation of No2 and So2 chemisorption on to nonpolar ZnO surfaces, Sensors and Actuators B: Chemical, 2009, 142, 179-184.
CrossRef - Duan, J., Huang, X., Wang, E., PEG-assisted synthesis of ZnO nanotubes, Material Letters, 2006, 60, 1918-1921.
CrossRef - Aslanzadeh, S., Transition metal-doped ZnO nanoclusters for carbon monoxide detection: DFT studies, J. Mol. Model, 2016, 22, 160.
CrossRef - Abdulsattar, M. A, Chlorine gas reaction with ZnO wurtzoid nanocrystals as a function of temperature: a DFT study, J. Mol, Model, 2017, 23, 125.
CrossRef - Patil, A. B., Patil, K. R., Pardeshi, S. K., Eco-friendly synthesis and solar photocatalytic activity of S-doped ZnO, J. Hazard. Mater., 2010, 183, 315.
CrossRef - Song, T. M., Wang, T. H., Li, J. C., First-principles study of periodic size-dependent band gap variation of Cu doped ZnO single wall nanotube, J. Mol. Model, 2012, 18, 5035.
CrossRef - Jianguang Wang, Li Ma, Yanhua Liang, Meiling Gao, Guanghou Wang, Density functional theory study of transition metals doped B80 fullerene, J. Theor. Comput. Chem, 2014, 13, 1450050.
CrossRef - Waranya Pipornpong, Benjawan Kaewruksa, Vithaya Ruangpornvisuti, DFT investigation on molecular structures of metal and non-metal doped ZnO sodalike cage and their electronic properties, Structural chemistry, 2016, 27, 773-784.
CrossRef - Mokhtar hjiri, Lasaad El Mir, Salvator Gianluca Leonardi, Micola Donato, and Giovanni Neri, Co and No2 selective monitoring by ZnO based sensors, Nano Materials, 2013, 3, 357-369.
- Chang Lin Yu, Kai Yang, Qingshu, Jimmy C. Yu, Fang Fang Cao, Xin Li, Xiaochun Zhou, Preparation, Characterization and photocatalytic performance of Mo-doped ZnO photocatalysts, Science China Chemistry, 2015, 55, 1802-1810.
- Swapna, R., Santhosh Kumar, M. C., Growth and Characterization of Mo-doped ZnO thin films by spray pyrolysis, Journal of Physics and Chemistry, 2013, 74, 418-425.
- Gokulakrishnan, V., Parthiban, S., Jeganathan, K., Ramamurthy, K., Investigation of Mo-doped ZnO thin films prepared by spray pyrolysis Technique, Ferroelectrics, 2011, 423, 126-134.
CrossRef - Ali Shokuhi Rad, Comparison of X12Y12 ( X=Al, B and Y=N, P) fullerene-like nanoclusters toward adsorption of dimethyl ether, J. Theor. Comput. Chem., 2018, 17, 1850013.
CrossRef - Becke A. D., Density functional exchange-energy approximation with correct asymptotic behavior, Physics Review A. 1988, 38, 3098-3100.
CrossRef - Lee, C., Yang, W., Parr, R. G., Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Physics Review B, 1988, 37, 785-789.
CrossRef - katarzyna pustule, Marcin Makowski, Assessing accuracy of exchange-correlation functional for electron affinities, J. Theor. Comput. Chem., 2017, 16, 1750052.
CrossRef - O’Boyle, N. M., Tenderholt, A. L., Langner, K. M., “Cclib: a library for package independent computational chemistry algorithm”, J. Comput. Chem., 2008, 9, 839-845.
CrossRef - Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Peterson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T., Montgomery, J. A., Jr, Peralta, J. E., Ogliaro, F, Bearpark, M., Heyd, J. J, Brothers, E., Kudin K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi M., Rega, N., Millam, M. J., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomeli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, C. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, O., Foresman, J. B., Ortiz, J. V., Cioslowski, J., Fox, D. J., 2009, Gaussian, Inc., Wallingford C. T., 2009, Gaussian 09, Revision D.01, Gaussian, Inc Wallingford CT, 2009.
- Ochterski, J. W., Thermochemistry in Gaussian Inc. Pittsburgh, PA, 2000.
- Manuel Alberto Flores Hidalgo, Diana Barraza Jimenez, Daniel glosman-Mitnik, Excited states analysis of sulfur substitutional impurities on (ZnO)6 cluster using DFT and TD-DFT, Journal of molecular structure: Theochem, 2010, 957, 100-107.
CrossRef - Ali Shokuhi Rad, Khurshid Ayub, Ni adsorption on Al12P12 nano cage: DFT study, J. Alloys. Comp. 2016, 678, 317-324.
CrossRef - Fatemeh Fallahpour, Milad Nouraliei, sara Soleimani Gorgani, Theoretical evaluation of a double functional heterogeneous nano sensors, Appl. Surf. Sci, 2016, 366, 545-551.
CrossRef - Saeed Aslanzadeh, Transition metal doped ZnO nanoclusters for carbon monoxide detection: DFT studies, J.Mol.Model, 2016, 22, 160.
CrossRef - Hamid Salimi, Ali Ahmadi Peyghan, Maziar Noel, Adsorption of Formic acid and Formate anion on ZnO nano cage: A DFT study, J. Clustr. Science, 2013, 26, 609-621.
CrossRef - Manuel Alberto Flores-Hidalgo, Diana Barraza-Jimenez, Daniel Glossman-Mitnik, Effects of sulphur substitutional impurities on (ZnO)n clusters (n=4-12) using density functional theory, Comput. Theor. Chem, 2011, 965, 154-162.
CrossRef - Nour El Houda Bensiradj, Amar Saal, Azeddine Dekhira, Quirda Quamerali, Theoretical study of selenium and tellurium impurities in (ZnO)6 clusters using DFT and TD-DFT, Int. J. Quantum Chemistry, 2016, 116, 1823-1898.
- Birajdar, S. D., Khirade, P. P., Bhagwat, V., Humbe, A. V., Jadhav, K., Synthesis, structural, morphological, optical and magnetic properties of Zn 1− x Co x O (0≤ x≤ 0.36) nanoparticles synthesized by sol gel-auto combustion method. Journal of Alloys and Compounds, 2016, 683, 513-526.
CrossRef - Seetawan, U., Jugsujinda, S., Seetawan, T., Ratchasin, A., Euvananont, C., Junin, C., Thanachayanont, C., Chainaronk, P., Effect of calcinations temperature on crystallography and nanoparticles in ZnO disk. Mater. Sci. Appln, 2011, 2, 1302–1307.
CrossRef - Zhong Lin Wang, nanostructures of ZnO, Materials today, 2004, 7, 26-33.
CrossRef - Samson R Woodley, Alexy, A., Sokol, Richard, C., Catlow, A., Abdullah, A., Al-Sunaidi, Scott M. Woodley, Journal of Physical Chemistry, 2013, 117, 27127-27145.
- Fatemeh Fallahpour Milad Nouraliei, Sara Soleimani Gorgani, Theoretical evaluation of Double-functional Heterogeneous Nano-Sensor, App. Surf. Sci, 2016, 366, 545-551.
CrossRef - Dheivamalar S, Sugi L, Density functional theory (DFT) investigation on doped fullerene with hetero atom substitution, Spectrochim. Acta Part A, 2015, 151, 687-695.
CrossRef - Anitha, Varughese, Characterization, thermal effect on optical band gap energy and photoluminescence in wurtzite ZnO: Er nanocrystallites, J. Material processing Tech, 2015, 3, 145-151.
- Wen Liu, Fan-Hua Meng, Jian- Hua Zhao, Xiao- Hui Jiang, A first principles study on the electronic transport properties of zig zag grapheme/grapheme nanoribbons, J. Theor. Compt. Chem., 2017, 16, 1750032.
CrossRef - Pal, S., Sarkar, A., Shallow acceptor state in ZnO realized by ion irradiation and annealing route, J. alloys and Compounds, 2017, 703, 26-33.
CrossRef - Abram, T., Chitra, S., Bejjit, L., Bouachrine, M., Lakhlifi, T., Electronic and photovoltaic properties of new materials based on 6-mono substituted and 3,6-disubstituted acridines and their application to design novel materials, for organic solar cells, J. Comp. Methods in Mol. Designs, 2014, 4, 19-27.
- Chandraboss, V. L., Karthikeyan, B., Senthilvelan, S., Experimental and first principle study of guanine adsorption on ZnO clusters, Phys. Chem. Chem. Phys, 2014, 16, 23461.
CrossRef - Koopmans, T., ordering of wave functions and eigen energies to the individual electrons of an atom, physica 1, 1933, 104-113
- Phool Singh Yadav, Dheeraj Kumar Pandey, A DFT study for the structural and electronic properties of ZnmSen nanoclusters, App.Nanosci., 2012, 2, 351-357.
CrossRef - Dheivamalar, S., Sugi, L., Ambigai, K., Density functional theory study of exohedral carbon atoms effect on electrophilicity of nicotine: Comparative analysis, Comp. Chem, 2016, 4, 17-31.
CrossRef - Masoud Bezi Javan, Alireza Soltani, Tazikeh Lemeski E, Afsaneh Ahmadi, Sahar Moazen Rad, Interaction of B12N12 nano cage with cysteine through various functionalities: A DFT study, Sup. Latt. Mic. Struc, 2016, 100, 24-37.
- Gaussview 4.1.2, Gaussian Inc., Wallingford, CT, 2004.
- Ali Shokuhi Rad, Khurshid Ayub, Detailed surface study of adsorbed nickel on Al12N12 nano cage, Thin solid films, 2016, 612, 179-185.
CrossRef - Agarwal, D. C., SHI induced modification of ZnO thin film: optical and structural studies, Nucl. Instrum. Methods Phys. Res., Sect. B, 2006, 244, 136-140.
- Wahab, R., Kim, Y. S., Hwang, I. H., Shin, H. S. A non aqueous synthesis, characterization of zinc oxide nano particles and their interaction with DNA, Synth. Met. 2009, 159, 2443-2452.
CrossRef - Deneva, I. M., Infrared spectroscopy investigation of metallic nano particles based on copper, cobalt, and nickel synthesized through borohydride reduction method, J. Univ. Chem. Technol. Metall., 2011, 45, 351-378.
- Sowbagya, Ananda, S., synthesis and characterization of Mo-doped ZnO nano particles by electrochemical method: Photodegradation kinetics of methyl violet dye and study of anti bacterial activities of MO- doped ZnO nano particles, International journal of nanomaterials and biostructures, 2015, 5, 7-14.
- Mishra, A., Bauerla, P., Small Molecule Organic Semiconductors on the Move: Promises for Future Solar Energy Technology, Angew. Chem. Int. Ed., 2012,51, 2020-2067.
CrossRef - Qin, C., Clark, A. E., DFT characterization of the optical and redox properties of natural pigments relevant to dye-sensitized solar cells, Chem.Phys.Let.,2007, 438, 26-30.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


