Synthesis and Evaluation of Central Antinociceptive Activity of Ring Substituted Chalcones; Molecular Docking Studies with Monoacylglycerol Lipase (MAGL) Enzyme
Shaheen Begum , Arifa Begum and Bharathi Koganti
, Arifa Begum and Bharathi Koganti
Institute of Pharmaceutical Technology, Sri Padmavati Mahila Visvavidyalayam (Women’s University), Tirupati 517502, Andhra Pradesh, India.
Corresponding Author E-mail: shaheen.pharmchem@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/3404024
Article Received on : 27-04-2018
Article Accepted on : 19-06-2018
Article Published : 24 Jul 2018
Chalcones possess Michael acceptor property due to the presence of α,β-unsaturated enone moiety in their structure. In the present study, molecular docking was performed to predict binding affinity of ring substituted chalcones with Monoacylglycerol lipase (MAGL), a serine hydrolase enzyme which can inhibited by Michael acceptors such as maleimide derivatives. 3, 4-Dimethoxy derivative, 3h, with -44.45 kJmol-1 of interaction energy, exhibited highest binding affinity and formed Pi-Sulphur interactions with methionine-123 residue of MAGL enzyme. As MAGL is an emerging target for antinociceptive drug development, ring substituted chalcones were synthesized and evaluated for their central antinociceptive activity using tail immersion and hot plate methods. The results revealed that compound 3h, chalcone bearing methoxy groups at 3rd and 4th positions of phenyl ring exhibited good antinociceptive activity in both the models. Good correlation was observed between antinociceptive activity and binding affinity toward MAGL in case of compound 3h.
KEYWORDS:Hot Plate Test; Monoacylglycerol Lipase; Molecular Docking; Ring Substituted Chalcones; Tail Immersion Test
Download this article as:| Copy the following to cite this article: Begum S, Begum A, Koganti B. Synthesis and Evaluation of Central Antinociceptive Activity of Ring Substituted Chalcones; Molecular Docking Studies with Monoacylglycerol Lipase (MAGL) Enzyme. Orient J Chem 2018;34(4). |
| Copy the following to cite this URL: Begum S, Begum A, Koganti B. Synthesis and Evaluation of Central Antinociceptive Activity of Ring Substituted Chalcones; Molecular Docking Studies with Monoacylglycerol Lipase (MAGL) Enzyme. Orient J Chem 2018;34(4). Available from: http://www.orientjchem.org/?p=47542 |
Introduction
Pain is now recognized as a global health problem, with an urge for effective and safer therapeutic agents. Pathophysiology of pain is complex despite of the recent advancements that has taken place in the area of molecular and neurobiology.1,2 Current findings suggest variety of drug targets including ion channels, playing pivotal role in progress of pain signalling [sodium, potassium and transient receptor potential (TRP) channels] and receptors involved in pain perception [imidazoline (I2), adenosine (A1Rs) and cholecystokinin (CCK)].3-5 Enzymes of the endocannabinoid cascade, especially, Fatty acid amide hydrolase (FAAH), α, α, β-Hydrolase domain containing 4,6,12 (ABHD-4, ABHD-6, ABHD-12) and Monoacylglycerol lipase (MAGL) which can hydrolyze endocannabonoids, 2-arachidonyl ethanolamine (AEA) and 2-arachnidonyl glycerol (2-AG) are among the promising pain targets and the compounds that alter their respective endocannabinoid levels have been represented as potential antinociceptive agents.6,7
Monoacyl glycerol lipase (MAGL), terminates pharmacological responses of 2-AG by hydrolysing it into arachidonic acid, a familiar pro-inflammatory mediator and glycerol.8 2-AG regulates various neurological and metabolic functions such as neuropathic pain, anxiety and neuromodulation by acting as a full agonist for the cannabinoid receptor type 1 (CB1) receptors.9 Hence, MAGL inhibition is a promising strategy to treat diseases in which higher 2-AG concentrations would be beneficial, in particular, pain and neuromodulation. MAGL is a member of serine hydrolase class of enzymes. It is highly expressed in brain, particularly at the presynaptic terminals and in areas of brain where CB1 receptors are localized.10,11 Synthetic drugs such as rivastigmine, saxagliptin, boceprevir, orlistat and β-lactam antibiotics are few examples of clinically approved inhibitors of human serine hydrolases.12,13 Literature survey revealed that MAGL enzyme can be inhibited by several chemical entities such as arylthioamides, carbamates, disulphide containing compounds and maleimides. These inhibitors act either by acting on catalytic nucleophilic amino acid Ser-122 (Ser-His-Asp triad) or by forming Michael adduct with sulfhydryl groups of aminoacids (Cys-204&Cys-242 present in the active site.14-16
Chalcones obtained either from natural sources or prepared synthetically, inhibit a broad range of enzymes, to mention a few, inducible nitric oxide synthase (iNOS), lipooxygenase (LOX), xanthine oxidase, tyrosine kinase, monoamine oxidase, soluble epoxide hydrolase, cyclooxygenase-2 (COX-2), angiotensin converting enzyme (ACE) and β-secreatase.17-22 Diverse set of therapeutic activities have been ascribed to chalcones such as antitumoral, anticancer, antioxidant, antifungal, antimitotic, chemoprotective, anti-inflammatory, antimicrobial, antinociceptive and antibacterial activities which invigorates interest of medicinal chemists. Presence of the, α,β-unsaturated enone unit in the chalcone is believed to be highly active, which impart enzyme inhibitory efficiency to this molecule. Michael-addition reaction occurs when chalcones interact with reactive groups of enzymes such as sulfhydryl functionality present in cysteine (Fig.1) which results in the loss of enzymatic activity.23-26
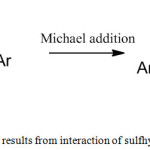 |
Figure 1: Enzyme inhibition results from interaction of sulfhydrul group and chalcone |
Chalcones derived from natural origin or synthetic chalcones bearing different substituent groups such as carboxylic acid or acetamide group or substituted urea exhibit peripheral as well as central antinociceptive activity. Mechanism of these potential molecules is not well defined. Literature indicates the probable mechanism might involve COX-2 and iNOS mediated pathways.27
In our earlier studies, we have performed molecular docking studies of ring substituted chalcones on iNOS and COX-2 enzymes and reported that these molecules have good binding affinity for both the enzymes, where electronic effects, hydrogen bonding ability have profound influence on the binding interactions. Molecular and pharmacokinetic properties prediction showed that these chalcones have good brain penetration permeability.28
In view of the capability of chalcones to interact with variety of enzymes and with the assumption that they may bind and form Michael adduct with cysteinyl groups of MAGL, it was proposed to dock a series of ring substituted (different electron releasing or withdrawing groups on ring B) chalcones with MAGL enzyme. Further, these chalcones were synthesized and their central antinociceptive activity was screened using the tail-immersion and hot-plate tests to find a correlation between in silico and in vivo studies.
Materials and Methods
Molecular Docking Studies
Molecular docking studies on MAGL enzyme was carried out on the crystal structure of MAGL which was retrieved from PDB data bank (PDB ID: 3PE6) using CDOCKER protocol, module of Discovery Studio Version 3.1 (Accelrys). CDOCKER is a more reliable grid-based protocol wherein CHARMM force fields are applied. While docking the molecules, 10 Å of grid extension was set around the active site of enzyme. Hydrogen atoms were added to the crystal structure of MAGL and all ionisable residues were set at pH 7. For each chalcone (Fig.2), ten best binding poses were generated. Input site sphere dimensions were set to -11.2454, 19.8898, -8.42488 and 11.7564 respectively and binding interactions were analysed. The molecular docking scores were calculated as binding free energies (kJ/mol).
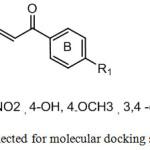 |
Figure 2: Chalcones selected for molecular docking studies |
Chemistry
Substituted acetophenones and benzaldehyde were purchased from Merck, India and Sigma-Aldrich, USA chemicals. All other chemicals are of AR grade. “Thermonik Precision Melting point cum Boiling point apparatus, Model C-PMB-2 was used to determine the melting points which were uncorrected. Formation of the final compounds was monitored by thin-layer chromatography (TLC). The infrared (IR) spectra were recorded using KBr pellets on Perkin-Elmer Spectrum-BX-I Infrared Spectrophotometer (cm-1). The 1H NMR spectra were recorded in deutero chloroform (CDCl3) on Jeol-400MHz instrument. Mass (m/z) spectra were obtained using LCMS-8030 mass spectrometer.
General procedure for the preparation of ring substituted chalcones (3a-j)
Chalcones were synthesized according to the previously described methodology.29 A substituted acetophenone (5 mmol) and benzaldehyde (5 mmol) were dissolved in dry methanol (30 ml). To this methanolic solution, 5ml of sodium hydroxide (50%) was slowly added. Stirring was continued (20 min -1 hr) at room temperature and to neutralize the contents, 1N HCl was added. Reaction time taken for the completion of reactions is mentioned in table.1. A mixture of ethyl alcohol and water was used for recrystallization. The synthetic procedure and reaction conditions for chalcones are illustrated in scheme 1.
Table 1: Reaction time taken for completion of different reactions
|
Compound No. |
R |
Reaction time |
|
3a |
4-H |
>45min |
|
3b-3d |
4-F,4-Cl, 2,5-(Cl)2 |
<30 min |
|
3e |
4-NO2 |
<20 min |
|
3f |
4-OH |
>50 min |
|
3g-3j |
4-OCH3, 3,4-(OCH3)2,3,4,5-(OCH3)3, 4-CH3 |
>40 min |
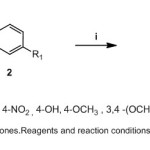 |
Scheme 1: Synthesis of chalcones Reagents and reaction conditions:(i) 50% NaOH, CH3 OH, room temperture |
Pharmacological Studies
The protocol used to perform the pharmacological activites was approved by institutional animal care and use committee ((1677/PO/a/12/IAEC/44). Male Swiss Albino mice (25-32 g) were used which were acclimatized to the laboratory for at least 2 h before testing. The test compounds were suspended well in 1% sodium carboxymethyl cellulose solution freshly before administration to animals. Tramodol was used as the standard (10mg/kg) drug which was administered orally to compare the results of the synthesized compounds.
Hot-Plate Method
This test was performed in order to measure the latency time (licking the hind- paws/ jumping off the hot plate) using hot-plate (30). The hot plate used in this study was set to a temperature of 55±1º C. The saline solution (0.1 ml/10g, p.o) was given to control animals whereas test group animals received synthesized compounds (50 mg/kg, p.o). To reduce the risk of tissue damage or injury, the cut-off time for the latency time was set at 20s.
Tail-Immersion Test
In this animal model, after oral administration of the chalcones or saline, mice were covered using cloth while their tails were immersed in a beaker of hot water (55 ± 1°C). The latency (seconds), for the animal to withdraw its tail from hot water was recorded. As long exposure to hot water may lead to tissue inflammation, a cut off of 15 sec was set.31
Results and Discussion
Chalcones containing different electron withdrawing and electron releasing substituent groups on various positions of B ring (3a-j) were subjected to molecular docking studies. Based on the results observed in these studies, we have synthesized the chalcones and screened them for their potential central antinociceptive activity using pain models such as tail immersion assay and hot-plate tests.
Molecular Docking Studies
MAGL enzyme belongs to a/b serine hydrolase super family and comprise of catalytic triad, nucleophilic elbow and oxyanion hole as the main structural elements. Catalytic triad constitutes the residues Ser-122, Asp-239 and His-269 in the the binding pocket. The catalytic site of MAGL is divided into two important regions; Leu-148, Ala-164, Leu-176, lle-179, lle- 211, Leu-213, Leu-214 and Val-217 side chains form hydrophobic pocket, while polar groups of Tyr-58, His-121and His-272 side chains, Glu- 53, Arg-57 and Ala- 51 form hydrophilic pocket.10-11
N-arachidonyl maleimide (NAM), a reported maleimide derivative (Michael acceptor) with potent MAGL inhibitory activity was used as standard inhibitor for comparing the molecular docking results of the synthesized chalcones. Maleimides acts as Michael acceptors and interact with cysteine residue of MAGL enzyme. Molecular docking studies of NAM with MAGL showed that the long aliphatic chain of the standard inhibitor was found to be located in a way that the terminal alkyl part connects with alkyl portions of Val-191 and Lys-278 via alkyl-alkyl interactions. The polar heterocyclic pyrrole ring of NAM showed π-π stacking interactions with Phe-159 where as the Pi electrons involve in Alkyl-Pi interactions with Leu-213 and Leu- 214 located near the active site. The interaction energy obtained for NAM was -60.98 kJmol-1 indicating its high affinity towards the enzyme (Fig.3).
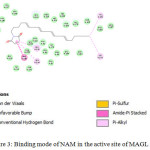 |
Figure 3: Binding mode of NAM in the active site of MAGL enzyme. |
Molecular docking studies of chalcones within the active site of MAGL revealed one particular observation with respect to all chalcones is the formation of S-π interactions (Sulphur-Pi) of both the phenyl rings of chalcone with crucial amino acid residue Met-123 residing adjacent to Ser-122, part of catalytic triad. Methionine side chains are both lipophilic, flexible and divalent sulphur behaves as a weak negatively polarized atom when interacts with phenyl rings. Ala-126, Ala-127 and Ala-216 were found to interact and have Pi-alkyl interactions with the phenyl rings of chalcones. Amide-Pi stacking interactions were also observed with His-121 and ring B which holds carbonyl group of α,β-unsaturated carbonyl system. These interactions were observed with all the docked chalcones irrespective of their substituent groups. Obtained results were different from our assumption that chalcones may bind and form Michael adduct with cysteinyl groups of MAGL.
When unsubstituted chalcone was docked in to the binding site of MAGL, Van der Waals interactions were observed between ring A and amino acid residue Ile-217 (-38.56 kJ/mol.). Introduction of fluorine increases binding affinity (-40.23 kJ/mol), fluorine forms a hydrogen bond with Val-217. Replacement of fluorine with chlorine resulted in further increase in binding affinity (-40.98 kJ/mol) indicating that presence of halogens are favourable for binding with the target protein MAGL.
Table 2: Docking scores for substituted chalcones (3a-j) with MAGL using CDocker (Discovery Studio)
|
Compound No. |
R |
Interaction Energy (kJ mol-1)a |
Interacting Amino acids |
|
3a |
4-H |
-38.56 |
Met-123 |
|
3b |
4-F |
-40.23 |
Met-123, Ala1-26 Ala-216 |
|
3c |
4-Cl |
-40.98 |
Met-123, Ala-126 Ala-216 |
|
3d |
2,5-(Cl)2 |
-39.24 |
Met-123, Ala-126 Ala-216 |
|
3e |
4-NO2 |
-41.43 |
Met-123, Ala-126 Ala-216 |
|
3f |
4-OH |
-40.40 |
Met-123, Ala-126Ala-216 |
|
3g |
4-OCH3 |
-43.23 |
Met-123, Ala-126 Ala-216 |
|
3h |
3,4-(OCH3)2 |
-44.45 |
Met-123, Ala-126Ala-216 |
|
3i |
3,4,5-(OCH3)3 |
-42.24 |
Met-123, Ala-26Ala-216 |
|
3j |
4-CH3 |
-37.43 |
Met-123, Ala-216, Ala126 |
|
NAM |
– |
-60.98 |
Phe-159,Val-191,Lys-273, Leu-123, Leu-124 |
a. The score represents the highest ranking poses obtained from the docking of each compound bound to MAGL enzyme.
Introduction of electron withdrawing group (-NO2) increased the binding affinity for the enzyme (-41.43 kJ/mol) when compared to unsubstituted chalcones (-38.56 kJ/mol). Among the methoxy containing chalcones, 3, 4-dimethoxy chalcone exhibited highest binding affinity with the enzyme (-44.45 kJ/mol) indicating the importance of substitution on chalcones. Methoxy hydrogens, situated at 3rd position of this chalcone found to form hydrogen bonding with Phe-209. Pi-donor hydrogen bonds were observed between phenyl rings of chalcones and backbone nitrogen atoms of amino acids Ala-126 and Ile-127(Fig.4).
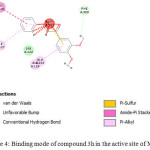 |
Figure 4: Binding mode of compound 3h in the active site of MAGL. |
The results obtained in the docking studies with MAGL indicate that synthesized chalcones have good affinity for this enzyme as they could interact with one of the important amino acid (Ser-122) present in the catalytic triad. Results demonstrated that substitution with either halogens, electron withdrawing and releasing groups are favourable for binding with the enzyme.
Chemistry
Ring substituted chalcones were prepared by condensation of the appropriate aromatic methyl ketone and simple benzaldehyde. After purification, they were obtained in moderate to good yields. The structures of title compounds were characterized spectral data (IR, 1H-NMR and Mass)
The IR spectra of the chalcones showed bands due to absorption in the regions 1658-1672 cm-1 (C=O str), 1596-1620 cm-1 (C=C str). In 1H-NMR spectra, the coupling constant for vinyl protons was found to be around 15.2-15.6 Hz, confirming the trans-configuration of the prepared chalcones. The aromatic protons showed signals around 7.0-7.9 ppm. The Characteristic molecular ion peaks were obtained [M+1]+ corresponding to the molecular weights of the title compounds. Spectral data including IR, 1H-NMR and Mass of the compounds were in good agreement with the literature values.26
The synthesized chalcones (3a-j) were tested for their central analgesic activities using hot plate test and tail immersion tests at a dose of 50 mg/kg body weight. Tramodol was used as standard drug for comparing the activity of chalcones.
Spectral data
Compound -3a
Chalcone [R=H]: IR ( νmax, cm–1): 1661 (C=O str), 1605 (C=C str);1H-NMR : 6.58-6.97 (d, 1H, CHα=C J=15.6 Hz), 7.11-7.50 (d, 1H, C=CHβ, J=15.6 Hz), 7.53–7.89 (m, 10H, Ar-H); MS: m/z 209.1 [M+1]
Compound -3b
3-(4-Fluorophenyl)-1-phenylprop-2-en-1-one [R=F]: IR ( νmax, cm–1): 1672 (C=O str), 1620 (C=C str);1H-NMR: 6.61-7.00 (d, 1H, CHα=C, J=15.6Hz), 7.15-7.54 (d, 1H, C=CHβ, J = 15.6Hz), 7.59–8.01 (m, 9H, Ar-H); MS: m/z 227 [M+1]
Compound -3c
3-(4-Chlorophenyl)-1-phenylprop-2-en-1-one [R=Cl]: IR ( νmax, cm–1): 1668 (C=O str), 1615 (C=C str);1H-NMR: 6.38-6.77 (d, 1H, CHα=C, J=15.6Hz ), 6.89-7.28 (d, 1H, CH=CH, J = 15.6), 7.38–7.76 (m, 9H, Ar-H); MS: m/z 243 [M+1].
Compound -3d
3-(2,5-Dichlorophenyl)-1-phenylprop-2-en-1-one [R=2,5-(Cl)2]: IR (νmax, cm–1): 1659 (C=O str), 1596 (C=C str);1H-NMR : 6.89 -7.27(d, 1H, CHα=C, J = 15.2 Hz), 7.36–8.12 (m, 9H,Ar-H& C=CHβ); MS: m/z 277 [M+1]
Compound -3e
3-(4-Nitrophenyl)-1-phenylprop-2-en-1-one [R=NO2]: IR ( νmax, cm–1): 1660 (C=O str), 1598 (C=C str);1H-NMR : 6.74-7.12 (d, 1H, CHα=C, J = 15.2 Hz), 7.25–7.86 (m, 10H, Ar-H& C=CHβ); MS: m/z 254 [M+1].
Compound -3f
3-(4-Hydroxyphenyl)-1-phenylprop-2-en-1-one [R=OH]: IR ( νmax, cm–1): 1674 (C=O str), 1615 (C=C str);1H-NMR : 5.34 (s,1H,OH) 6.78-7.17 (d, 1H, CHα=C, J=15.6), 7.25–7.76 (m, 10 H , Ar-H& C=CHβ); MS: m/z 225 [M+1]
Compound 3g
3-(4-Methoxyphenyl)-1-phenylprop-2-en-1-one [R=4-OCH3]: IR ( νmax, cm–1): 1671 (C=O str), 1605 (C=C str);1H-NMR: 3.01(s,3H,OCH3) 6.80-7.19 (d, 1H, CHα=C, J=15.6Hz), 7.25-7.76 (m,10 H, Ar-H& C=CHβ); MS: m/z 239 [M+1].
Compound -3h
3-(3,4-Dimethoxyphenyl)-1-phenylprop-2-en-1-one [R=3,4-(OCH3)2]: IR (νmax, cm–1): 1665 (C=O str), 1596 (C=C str); 1H-NMR : 3.05 (s,6H,(OCH3)2) 6.78-7.17 (d, 1H, CHα=C, J=15.6Hz ), 7.25–7.76 (m, 10 H,Ar-H& C=CHβ); MS: m/z 269 [M+1].
Compound -3i
1-Phenyl-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one [R=CH3]: IR ( νmax, cm–1): 1658 (C=O str), 1621 (C=C str); 1H-NMR : 3.00 (s,6H,(OCH3)2) 3.04(s,3H,OCH3) 6.67-7.06 (d, 1H, CHα=C, J=15.6Hz), 7.25–7.95 (m, 10 H, Ar-H& C=CHβ); MS: m/z 223 [M+1]
Compound -3j
1-Phenyl-3-p-tolylprop-2-en-1-one [R=CH3]: FT-IR ( νmax, cm–1): 1660 (-C=O), 1606 (-C=C-);1H-NMR : 1.12 (s,3H,CH3), 6.38-6.77 (d, 1H, CHα=C J=15.2Hz ), 7.25–7.76 (m, 10 H,Ar-H, & C=CHβ); MS: m/z 223 [M+1]
Central Antinociceptive Activity
Ring substituted chalcones displayed peripheral antinociceptive activity in acetic acid induced writhing assay.26 To evaluate their central antinociceptive activity, tail immersion and hot plate methods were used.
Tail Immersion Test
Results of the tail immersion assay showed that most of the chalcones were active in this test. The oral administration of compound 3g and 3h significantly increased the latency time (9.3±0.22 & 9.6±0.28 sec) to the nociceptive response in this test comparable to that of standard drug tramodol (11.5±0.32 sec). Unsubstituted chalcone, 3a, was found to be moderately active in this assay (6.01±0.62 sec). Introduction of halogens such as fluorine and chlorine improved the activity (7.0±0.22 &7.5±0.30 sec). Halogen containing chalcones 3b, 3c and 3d showed good activity in this assay suggesting that presence of this halogen atom is favourable for central antinociceptive activity.
Presence of hydroxyl group on chalcone was unable to exert any promising activity in this assay (5.01±0.20 sec) whereas replacement of hydroxy group with methoxy functionality (3f) significantly improved the central antinociceptive with latency period of 6.52±0.67 sec. Results of this assay indicates ability of chalcones to act as central antinociceptive agents.
Hot Plate Test
Profile of antinociceptive activity of chalcones in this assay showed that 3h showed increased latency period up to 8.0±0.25sec which is almost comparable to latency of tramodol, 8.3±0.21sec. The next chalcone which was active in this test was 3i, 3, 4, 5-trimethoxy chalcone, which had the latency of 7.2±0.25 sec. The results demonstrate that halogen containing chalcones, 3b, 3c and 3d, which were moderately active in tail immersion assay were found to be moderately active even in hot plate test.
The hot-plate test measures supraspinal responses and the tail-immersion test measures spinal level responses. Hence combined, they represent good models for assessing the central level antinociceptive activity.31-33 Chalcones showed marked activities in the hot-plate and in the tail-immersion tests (Table. 3), suggesting that they were able to cross blood brain barrier and active on the central nociceptive pathways.
Table 3: Antinociceptive activities of substituted chalcones (3a-j) in different central antinociceptive models
| S.No. | R | Tail immersion test(Latency period in sec) | Hot-Plate test(Latency period in sec) |
|
Control |
– |
3.2±0.16 |
2.5±0.22 |
|
Tramadol |
– |
11.5±0.22* |
8.3±0.21 |
|
3a |
4-H |
6.01±0.62* |
3.6±0.21* |
|
3b |
4-F |
7.0±0.22* |
5.3±0.21* |
|
3c |
4-Cl |
7.5±0.30* |
5.3±0.21* |
|
3d |
2,5-(Cl)2 |
7.5±0.30* |
4.3±0.22 |
|
3e |
4-NO2 |
6.01±0.62 |
3.6±0.21* |
|
3f |
4-OH |
5.01±0.20* |
3.8±0.22* |
|
3g |
4-OCH3 |
9.3±0.22 * |
5.3±0.24* |
|
3h |
3,4-(OCH3)2 |
9.6±0.28* |
8.0±0.25* |
|
3i |
3,4,5-(OCH3)3 |
7.5±0.30 |
7.2±0.25 |
|
3j |
4-CH3 |
3.2±0.16*s |
3.8±0.22* |
Values are expressed as Mean± SEM, Values represent one-way analysis of variance (ANOVA) followed by Dunnett`s test. *p<0.05 Vs control (n=number of animals in each group=6)
Conclusion
Chalcones, the small organic molecules possess good binding affinity for MAGL enzyme, derivative 3h bearing methoxy group at both 3rd and 4th positions showed highest affinity. Chalcones possessing halogens 3b, 3c, 3d and methoxy groups 3f, 3g and 3h exhibited good central analgesic activity in tail immersion and hot plate methods and highest activity was observed with derivative 3g, which showed high affinity for MAGL enzyme.
Acknowledgements
We are grateful to DST-CURIE (Department of Science & Technology- Consolidation of University Research for Innovation and Excellence in Women Universities), Sri Padmavathi Mahila University, for providing FTIR spectrum.
References
- Borsook, D.; Hargreaves, R.; Bountra, C.; Porreca, F. Sci. Transl. Med. 2014, 6 (249): 249
CrossRef - Lee, G.H.; Kim, S.S. Mediators Inflamm. 2016, Article ID 5808215.
- Li, J.X.; Zhang, Y. Eur. J. pharmacol. 2012, 681(1-3) : 1-5
CrossRef - Crow, M.; Denk, F.; McMahon S.B. Genome Med. 2013, 5: 12-22.
CrossRef - Yaksh, T.L.; Sarah, A.W.; Roshni, R.; Linda, S.S. F1000Prime Rep. 2015, 7:56.
CrossRef - Di-Marzo, V. Pharmacol Research 2009, 60: 77–84.
CrossRef - Muccioli, G.G. Drug. Discov. Today. 2010, 15 (11/12): 474-483.
CrossRef - Labar, G.; Wouters, J.; Lambert, D.M. Curr. Med. Chem. 2010, 17(24): 2588-2607.
CrossRef - Mulvihill, M.M.; Nomura, D.K. Life. Sci. 2013, 92(8-9): 492–497.
CrossRef - Bertrand, T.; Auge, F.; Houtmannn, J.; Rak, K.; Vallee, F.; Mikol, V.; Berne, P.F.; Michot, N.; Cheuret, D.; Hoornaert, C.; Mathieu, M. J. Mol. Biol. 2010, 396: 663-673.
CrossRef - Labar, G.; Bauvois, C.; Borel, F.; Ferrer, J.L.; Wouters, J.; Lambert, D.M. Chem. Bio. Chem. 2010, 11: 218 – 227.
CrossRef - Jonathan, Z.L.; Cravatt, B.F. Chem. Rev. 2011, 111(10): 6022–6063.
CrossRef - Bachovchin, D.A.; Cravatt, B.F. Nat. Rev. Drug. Discov. 2013, 11(1): 52-68.
CrossRef - Matuszak, N.; Muccioli, G.G.; Labar, G.; Lambert, D.M. J. Med. Chem. 2009, 52(23): 7410–7420.
CrossRef - Minkkila, A.; Saario, S.M.; Nevalainen, T. Curr. Top. Med. Chem. 2010, 10: 828-858.
CrossRef - Saario, S.M.; Laitinen, J.T. Basic. Clin. Pharmacol. Toxicol. 2007, 101: 287-293.
CrossRef - Rojas, J.; Dominguez, J.N.; Charris, J.E.; Lobo, G.; Paya, M.; Ferrandiz, M.L. Eur. J. Med. Chem. 2002, 37: 699–705.
CrossRef - Campos-Buzzi, F.; Padaratz, P.; Meira, A.V.; Correa, R.; Nunes, R.J.; Cachinel-Filho, V. Molecules. 2007, 12: 896-906.
CrossRef - Reddy, N.P.; Aparoy, P.; Reddy, T.C.; Achari, C.; Sridhar, P.R.; Reddanna, P. Bioorg. Med. Chem. 2010, 18 (16): 5807-15.
CrossRef - Kong, L.D.; Zhang, Y.; Pan, X.; Tan, R.X.; Cheng, C.H. Cell. Mol. Life. Sci. 2000, 57(3): 500-505.
CrossRef - Nerya, O.; Musa, R.; Khatib, S.; Tamir, S.; Vaya, J. Phytochemistry. 2004, 65(10): 1389-1395
CrossRef - Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yanez, M.; Orallo, F.; Ortuso, F.; Alcaro, S. J. Med. Chem. 2009, 52(9): 2818-2824.
CrossRef - Mesenzani, O.; Massarotti, A.; Giustiniano, M.; Pirali, T.; Bevilacqua, V.; Caldarelli, A.; Canonico, P.; Sorba, G.; Novellino, E.; Genazzani, A.A.; Tron, G.C. Bioorg. Med. Chem. Lett. 2011, 21: 764–768
CrossRef - Padhye, S.; Ahmad, A.; Oswal, N.; Sarkar, F.H. J. Hematol. Oncol. 2009, 2:38
CrossRef - Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chem. Rev. 2017, 117 (12):7762-7810
CrossRef - Correa, R.; Pereira, M.A.S.; Buffon, D.; Santos, L.; Cechinel Filho, V.; Santos, A.R.S.; Nunes, R. J. Archiv. der Pharmazie. 2001, 334 (10): 332-334.
CrossRef - Begum, S; Bharathi, K.; Prasad, K.V.S.R.G. J Global Trends Pharm Sci. 2016, 7(3): 3353 – 3361.
- Begum, S; Bharathi, K.; Mallika, A. International Journal of Pharmacy and Biological Sciences. 2016, 6 (2): 100-107.
- Li, R.; Kenyon, G.L.; Cohen, F.E.; Chen, X.; Gong, B.; Dominguez, J.N.; Davidson, E.; Kurzban, G.; Miller, R.E.; Nuzum, E.O. J. Med. Chem. 1995, 38: 5031–5037.
CrossRef - Eddy, N.B.; Leimback, D. Jour. Pharmacol. Exp. Ther. 1953, 107: 385-393.
- Lingadurai, S.; Nath, L.K.; Karl, P.K.; Besra, S.E.; Joseph, R.V. Afr. J. Tradit. Complement .Altern. Med. 2007, 4:411–416
CrossRef - Gabra, B.H.; Sirois, P. Peptides. 2003, 24(8):1131-1139.
CrossRef - Soujanya, M.; Rajitha, G. Asian. J. Chem. 2017, 29 (11): 2479-2484.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


