Applications of Computer-Aided Approaches to Determine Urease Inhibitory Activities of Flavonoids Analogous
Sheetal Gupta , Amrital V. Bajaj and Neena Sohani
, Amrital V. Bajaj and Neena Sohani
School of Chemical Sciences, Devi Ahilya University, Indore, Madhya Pradesh, India.
Corresponding Author E-mail: sheetalgb2b@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/3404040
Article Received on : 09-05-2018
Article Accepted on : 13-06-2018
Article Published : 03 Aug 2018
The objective of this study was to determine potency of newly synthesized flavonoids ligands against urease enzyme, which take place through strong bond formation between ligands and amino acid of the active site of urease and both metal ions [Ni (II)]. In order to correctly valuate ligands, molecular dynamic simulation was used. Simulation studies revealed scientific information such as perfect bond contribution, percentage contribution of bonds and stability of bonds with variable time length. Afterwards, the analysis of binding free energy and complex stability has been done through the molecular mechanics generalized Born surface area continuum solvation (MM/GBSA) method. Then, the root mean square deviation (RMSD) was used as post-docking scoring approach. Interestingly, compound number 28 was found to be the most potent candidate in terms of anti-urease activity. Study also suggested that further modification of base ligands with electronegative substituents could enhance potency of the potential drug candidates.
KEYWORDS:Binding Free Energy Analysis; Docking; Molecular Dynamic Simulation; RMSD
Download this article as:| Copy the following to cite this article: Gupta S, Bajaj A. V, Sohani N. Applications of Computer-Aided Approaches to Determine Urease Inhibitory Activities of Flavonoids Analogous. Orient J Chem 2018;34(4). |
| Copy the following to cite this URL: Gupta S, Bajaj A. V, Sohani N. Applications of Computer-Aided Approaches to Determine Urease Inhibitory Activities of Flavonoids Analogous. Orient J Chem 2018;34(4). Available from: http://www.orientjchem.org/?p=48059 |
Introduction
Since the early 1900s, various studies of flavonoid compounds have been conducted. Flavonoid compounds are a large class of compounds originally derived from plants, all of which share a similar chemical structure based on the flavone backbone. The flavone backbone is a tricyclic, polyphenolic organic structure comprised of a 15-carbon skeleton structure. Structurally flavonoid is made up of two cycles and 1 heterocyclic ring which are said to be the A, B and C rings, respectively, where A and B are conjugated benzene rings and C is a pyran ring.1 Alternatively, these structures are sometimes referred to as bioflavonoids.2 Flavonoids are widely distributed in plants and are responsible for a variety of red, yellow, and blue plant pigments found in flowers, barks, leaves, fruits, and roots, etc.3 Functionally, flavonoids are involved in processes such as UV-filtration, symbiotic nitrogen fixation, floral pigmentation for the attraction of pollinator animals, as well as integral components in signal transduction pathways, physicochemical regulation, and cell cycle inhibition. In non-plant species, these compounds play an important role in digestion, nutrient absorption, and other metabolic processes.4-8 Almost 5000 varieties of flavonoid compounds have been derived and identified.9 Most of these varieties can be divided into 6 classes: anthoxanthins, isoflavones, anthocyanins, flavanones, flavanonols and flavans.10-12 One of the most abundant flavonoids is Quercetin.13 Many derivatives of quercetin have been identified and characterized since it was named in 1857. Flavonoids have been identified in a variety of foods and are present in dark chocolates, red wine, green and black tea, banana, onion, citrus fruits and parsley, etc. In December 2013 a large database was created by the USDA, through which 506 food items were selected as “best flavonoid-containing foods”.14 Flavonols comprise the most widespread part of human diet of all classes of flavonoids.
Normally, bioavailability of flavonoids has been found to be quite low due to limited absorptions, extensive metabolism and breakdown in the body, and rapid excretion. Flavonoids have been shown to exhibit many biological and pharmacological activities and properties such as antiasthmatic, antitussive, antiabortive, antiparasitic, antipsoriatic, anti-acne, antiseborrheic, antioxidant, antiperuricemic, antiepileptic, antimigraine, anti-Parkinson, anti-infective, antibacterial, antiviral, and antimycotic functions.15-16 Additionally, these compounds are important dietary micronutrients. They are important in metabolizing vitamins A and D, sugars, and amino acids. Industrially they may be used in printed circuits, encapsulation, mountains, lipid and reflecting filters, and semiconductors, etc.
Materials and Methods
All MD simulations in this study were performed using Schrödinger (Schrödinger, LLC, and New York-2017-1) and various modules therein. Chemsketch was used to generate images of the two-dimensional (2D) structures.
Dataset Selection and Biological Activity
The dataset for the present study was screened as per the method outlined by Zhu-Ping Xiao et.al.in 2013.17 The dataset selected for modeling in this study consisted of a series of 30 Flavonoid derivatives that are known to present urease inhibitory activity.
 |
Table 1: Structure of series of flavonoid analogous along with their biological activity. |
The chemical structure and biological activity (IC50) for each of the studied molecules is shown in Table 1. Though all 30 molecules exhibited urease inhibitory activity, molecules 13, 15, 21, 14, 2 and 9 were found to be particularly efficacious. Upon further analysis, it is apparent that the efficacy of a molecule as a urease inhibitor is predominantly dependent upon the substituent present at the aromatic residue, whereas variation in the substitution pattern of the aromatic part of the molecule has little effect.
Molecular docking
Detailed descriptions of the methods utilized for this assessment are given in the following sections.
Ligand Preparation
The 2D structures of the studied thiobarbituric acid derivatives, considered here to be ligands, were prepared using the Lig Prep application in the Schrödinger Maestro Suite 2017-1 (Lig Prep, Schrödinger, 2017-1).18 The Lig Prep application optimizes the ligand structures upon conversion from 2D to 3D, corrects inaccurate bond lengths and bond orders, generates ionization states, and minimizes the structure’s energy.19 The structures prepared with Lig Prep were then utilized in docking simulations.
Protein Preparation and Grid Formation
The crystal structures of urease 1E9Y, 4AC7 and 4UBP were retrieved from the Protein Data Bank 20 and subsequently prepared using the Protein Preparation Wizard, which is accessible in the Schrödinger Suite 2017-1. Crystallographic water molecules – i.e. water molecules bound by less than three hydrogen-bonds – were removed, missing side-chain atoms were inserted, and hydrogen-bonds were added where appropriate, taking into consideration the ionization states at pH 7.0 for both the acidic and basic amino acid residues. After these corrections, Prime (Prime, Schrödinger 2017-1) 21 was utilized to reconstruct any remaining structural discontinuities. Energy minimization of the crystal structure was carried out using the OPLS_2005 force field. The radial region within 10 Å of the ligand in the complexed structure was designated as the enzyme’s active site. A grid box was then generated by selecting the crystalized ligand for docking purposes. To test the docking parameters, this catalytic cavity was used as the docking site for the lowest energy conformations of the studied compounds. This test was performed using the Grid-Based Ligand Docking with Energetics (Glide, Schrödinger 2017-1) 22 in ‘extra-precision’ mode. No constraints were applied at this stage.
Glide Docking
Glide works by internally producing and progressively filtering molecular conformations. In standard docking, the filters first reject any non-active molecules, then score molecules based on orientation, distance, hydrophobic contact with the ligand, hydrogen-bonding associations, and so on. This XP technique was used to reduce the occurrence of false positives and enhance the association between superior poses and high scores.
Free Energy Calculation
The free energy of binding (ΔGbind) was computed with a Prime/MM-GB/SA approach, which was used to predict the ΔGbind for each ligand-receptor pair. The energy of the docked pose for each complex was minimized via the nearby enhancement highlight tool in Prime, while the energy of each complex was calculated with the OPLS_2005 force field and the VSGB solvation model.23 ΔGbind 24 can be thought of as the sum of the change in minimized energy (ΔE), the change in energy of solvation (ΔGsolv), and the change in surface area energy (ΔGSA):
ΔGbind = ΔE + ΔGsolv + ΔGSA
ΔE, ΔGsolv, and ΔGSA can in turn be obtained by subtracting corresponding values for the free protein and ligand from that of the protein-ligand complex.
ΔE + Ecomplex – Eprotein – Eligand
ΔGsolv = ΔGsolv(complex) –Gsolv(protein) – Gsolv(ligand)
ΔGSA = GSA(complex)–GSA(protein) – GSA(ligand)
In this study, simulations were performed with the GBSA continuum model in Prime, which uses a surface-generalized Born (SGB) model.
MD Simulation
The molecular dynamic simulations were performed between 1E9Y and each ligand by using the Desmond software suite 25 and the OPLS 2005 force field. The system was then solvated by use of the internal system builder panel with the TIP-3P solvent model. Throughout the solvation process, the structure was kept 10 Å from the edges of the right prism-shaped box. In addition, the volume of the system was minimized such that the box size became 91550. A 20 Å region was excluded during the ion selection process; hence no ions or salts were deposited in that region. To ensure that the system could be neutralized, the salt concentration was set at 0.15M Na+/Cl—. The POPC membrane model was then used at 300K to establish the automatic membrane. After this process, the system reached equilibrium with the default values for temperature, ensemble class NTP, Berendsen thermostats, and Buro states. Finally, the model was relaxed using the RESPA integrator panel set at 2fs. All other panels were set to their default parameters. Bonded and non- bonded interactions were calculated, which yielded the final results.
Results and Discussions
Docking
Docking studies were performed in order to determine the effective and resultant interactions between protein and flavonoid-derived ligands. Information obtained from the docking study includes total number of hydrogen and other bonds formed in complex, and the distances of all these bonds. Additionally, information on compound compatibility with protein was obtained for all compounds in this series. This information was obtained for three proteins imported from PDB: 1E9Y, 4AC7 and 4UBP and given in Table 2.
Table 2: Comparative docking scores of different proteins
| Compounds | 1E9Ya | 4UBPb | 4AC7c |
| Docking Score | Docking Score | Docking Score | |
| 28 | -6.49 | -5.39 | -5.68 |
| 27 | -6.23 | -4.89 | -4.64 |
| 21 | -6.33 | -6.29 | -6.09 |
| 2 | -5.58 | -5.93 | -6.07 |
| 15 | -5.24 | -5.12 | -5.12 |
| 13 | -4.46 | -4.5 | -4.21 |
abc Protein data bank codes for Proteins
Protein 1E9Y was selected for docking of all compounds of the reported series. All results (such as hydrogen bonding, interacting amino acids, RMSD, and docking score) for this study are given in Table 3. Out of 30 compounds only 20 compounds were obtained through docking and rest of compounds were automatically discarded by Glide.
Compound 28 (Figure 1-2) was elected for further study based on its superior dock score. Compound 28 (common name luteolin) enters the protein active site city with the phenolic ring of oriented “headfirst” towards the cavity interior.
Inside the cavity, a dense hydrophilic, unspecified residues and polar reason were observed. These features create a favourable environment for the uptake and binding of ligands. In compound 28, both hydroxyl groups (-OH) attached to the phenolic ring form hydrogen bonds with amino acids residues located on the interior of the cavity. The meta- hydroxyl forms a hydrogen bond with Ala365 and para- hydroxyl bonds with Gly27. Compound 28 enters the cavity vertically oriented, but the other phenolic ring is unable to completely enter the cavity, and is instead oriented jutting outwards through the opening/mouth of the active site.
Table 3: Extra Precision Glide Docking Results with Interacting amino acids with 1E9Y
| Compounds | docking score | XP GScorea | glide energy | glide emodel | RMSD | H Bondsb | Interacting Amino Acids |
| 1 | -5.05 | -5.056 | -34.828 | -47.61 | 1.02 | 2 | Asp223, Cys321 |
| 2 | -5.796 | -5.816 | -31.056 | -35.389 | 2.13 | 3 | Val320, Gln364, Ala365 |
| 4 | -3.217 | -3.217 | -38.46 | -49.603 | 1.9 | 0 | His322 |
| 6 | -3.941 | -4.002 | -33.672 | -40.044 | 2.35 | 1 | His221, His322,Hie248 |
| 7 | -3.538 | -3.591 | -34.976 | -44.533 | 1.97 | 1 | Asp223 |
| 9 | -2.466 | -2.466 | -26.436 | -30.79 | 0.14 | 1 | His322, Asp362 |
| 13 | -4.493 | -4.509 | -31.165 | -39.281 | 0.91 | 2 | Ala365, Gly279 |
| 14 | -5.581 | -5.584 | -30.815 | -41.034 | 1.17 | 3 | His322 (3),Gly279, Ala365 (2) |
| 15 | -5.276 | -5.276 | -35.828 | -47.608 | 1.87 | 2 | Gly279, Asp223 |
| 17 | -4.081 | -4.143 | -37.908 | -46.41 | 1.89 | 1 | Arg338, His322 |
| 18 | -4.547 | -4.673 | -37.765 | -47.726 | 1.98 | 2 | His322, Asp223, Arg338 |
| 19 | -3.938 | -3.94 | -31.028 | -38.188 | 1.5 | 2 | Asp223, Ala365 |
| 20 | -4.723 | -4.739 | -37.806 | -47.611 | 0.76 | 4 | Asp165, Asp223, Arg338, Ala365 |
| 21 | -6.099 | -6.111 | -37.39 | -50.032 | 1.73 | 2 | Asp165,Gly279(2) |
| 25 | -4.453 | -4.461 | -29.973 | -38.846 | 0.5 | 1 | Asp223 |
| 26 | -5.224 | -5.241 | -34.961 | -46.814 | 0.72 | 3 | Asp165, Asp223, Gly279 |
| 27 | -6.212 | -6.234 | -35.926 | -46.515 | 0.62 | 4 | His322, Arg338(2),Gly279, Ala365 |
| 28 | -6.474 | -6.491 | -35.99 | -49.033 | 2.08 | 2 | Gly279, Ala365 |
| 29 | -3.524 | -3.524 | -31.159 | -41.606 | 1.13 | 0 | NIL |
| 30 | -4.081 | -4.099 | -28.421 | -40.746 | 0.24 | 2 | Gly279, Hie221, His322, Glu222 |
aExtra Precision Glide score
bHydrogen bonds
Compound 27 showed the second highest dock score of all scored compounds. The compound is based on the molecular structure of a flavonoid known by the common name baicalein. Its phenolic ring adjacent to the pyrone ring enters into the cavity and appears to be completely embedded in the cavity interior. All the three hydroxyl (-OH) substituted groups of the ligand are observed to participate in bonding between hydrophilic (Ala365), charged(Arg338), and polar(His322) amino acid residues as well as with water molecules (Gly279). Specifically, the meta- hydroxyl group forms a hydrogen bond with Arg338, the other m-OH group pairs with Ala365. Two hydrogen bonds form between p-OH Arg338 and Gly279, respectively.
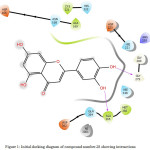 |
Figure 1: Initial docking diagram of compound number 28 showing interactions. |
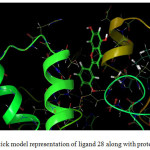 |
Figure 2: Ball and stick model representation of ligand 28 along with protein 1E9Y. |
Additional stability is lent to the structure due to the formation of a pi-pi stack between the pyrone ring and residue His322. The RMSD value for this complex was found to be very low (0.62), thus this compound shows least deviation during the docking event. Although the experimental IC50 value of this compound is quite high, formed bonds observed during simulation could be indicative of promising potential as a bioactive compound.
Compound 21 showed the next highest dock score. According to the experimental data compound 21 bioactivity is superior to that of compounds 28 and 27.
Compound 21 enters the cavity in a vertical orientation. Compound 13 is the compound which is experimentally best active. Compound 13 forms two hydrogen bonds between phenolic hydroxyl groups and amino acid residues: para- hydroxyl bonds with Gly279, and meta- hydroxyl bonds with Ala365. Thus, all compounds were shown to have somewhat favourable docking scores and hydrogen bonding, which indicate possible enhanced potency. In order to obtain more accurate results, docking studies were performed.
Simulation
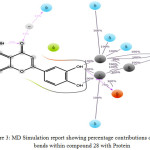 |
Figure 3: MD Simulation report showing percentage contributions of different bonds within compound 28 with. |
Protein
In Compound 28 (Figure 3-4) there was no significant correlation observed between experimental data and docking score. In the starting snapshot of the simulation, several bonds were observed between the ligand and various amino acid residues such as Asn168, His274, Ala365, His136, His138, etc. Some bonds when observed until the final snapshot were highly preserved: two hydrogen bonds between phenolic hydroxyl groups were preserved up to 95% and 100%. Additionally, the meta- hydroxyl group formed three bonds, two of which were with Ni (I) and Ni (II) metallic ions, preserved to 100%, and the third with Gly222 preserved up to 95%. Para- hydroxyl group formed one bond with Ni metal which was preserved to 100% in selected trajectories. Here, some intra- type ionic bonds were also observed, which were found to support ligand fit inside the active site cavity. Oxygen of the pyrone ring formed a bond with amino acid residue Gln364 preserved up to 33%. This bond formed through a water bridge. Additionally, the oxygen involved in this binding event was shown to participate in the formation of an intra-type bond with a phenolic hydroxyl group, which was preserved up to 33%. These results show that at the time of docking there were several complementary fit arrangements. Results were found to be in support of the docking score.
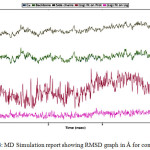 |
Figure 4: MD Simulation report showing RMSD graph in Å for compound 28. |
Among the selected compounds the second best bonding was observed in compound 21. In the starting snapshot, amino acid residues such as Ala278, Pro302, Glu311, Glu313, Leu318, etc. were observed to participate in bonding. The hydroxyl group of the phenolic ring bonded with residue Arg338, preserved up to 91%. Additionally, the phenolic ring formed a bond via pi-pi stacking with His314, preserved up to 43% in the selected trajectory. Analysis of compound 27, which showed the second best dock score, showed some favourable bonding events, though slightly less preserved. A hydrogen bond through Water Bridge with Glu311 amino acid residue formed with a phenolic hydroxyl group, preserved up to 37%. Intra-type bonding was also preserved in between a phenolic hydroxyl group and pyrone oxygen atom up to 41 % in the trajectory. Compound 13 which experimentally showed the best bioactivity showed underwhelming docking results during docking. However, during simulation many hydrogen bonds were observed. The phenolic meta- hydroxyl group formed a bond with residue Gly279, preserved to 63%. The para- hydroxyl group formed a bond with Thr251, preserved up to 35%. The oxygen of the pyrone ring formed a bond with Arg338 through Water Bridge preserved up to 36%. Similarly, compound 15 formed two hydrogen bonds between the para- hydroxyl group and Arg338 (through Water Bridge) and Ala275, preserved up to 45% and 38%, respectively.
MM-GB/SA
Table 4: Binding free energy calculation results for the different series of compound bound with 1E9Y All energies are in cal mol-1
| Compounds | ∆Gcoulomba | ∆GCovalentb | ∆GsolLipoc | ∆SolvGBd | ∆GvdWe | ∆GBINDf |
| 1 | -23.95 | 2.905 | -5.476 | 39.862 | -33.372 | -23.584 |
| 2 | -30.753 | 2.599 | -6.69 | 38.744 | -27.234 | -25.912 |
| 4 | -12.641 | 1.177 | -11.62 | 33.781 | -39.251 | -30.749 |
| 6 | -22.405 | 6.781 | -8.038 | 50.163 | -30.516 | -8.308 |
| 7 | -8.158 | 2.928 | -8.331 | 27.732 | -29.966 | -17.966 |
| 9 | -26.17 | 3.193 | -10.833 | 54.253 | -25.888 | -9.216 |
| 10 | -28.241 | 3.775 | -6.633 | 52.128 | -33.932 | -18.397 |
| 13 | -18.538 | 0.465 | -9.824 | 40.281 | -28.285 | -20.865 |
| 14 | -21.659 | 1.817 | -8.513 | 39.394 | -26.338 | -20.987 |
| 15 | -37.572 | 5.603 | -8.409 | 51.401 | -30.206 | -21.897 |
| 17 | -17.024 | 4.631 | -8.572 | 36.841 | -35.219 | -22.091 |
| 18 | -19.664 | 1.783 | -8.015 | 35.024 | -32.422 | -26.864 |
| 19 | -21.335 | 2.722 | -6.423 | 42.42 | -28.199 | -15.055 |
| 21 | -31.903 | -0.016 | -8 | 38.671 | -31.434 | -37.403 |
| 25 | -17.705 | 2.492 | -9.68 | 37.208 | -31.441 | -22.023 |
| 26 | -20.942 | 4.048 | -8.726 | 40.196 | -33.225 | -24.444 |
| 27 | -22.226 | 2.25 | -7.744 | 41.36 | -31.728 | -23.608 |
| 28 | -27.938 | 1.854 | -7.236 | 38.078 | -30.368 | -30.042 |
| 29 | -6.32 | 2.884 | -9.508 | 30.114 | -34.337 | -19.03 |
| 30 | 0.061 | 0.269 | -6.016 | 41.917 | -30.202 | 0.001 |
a Contribution to the free energy of binding from the Coulomb energy.
b Contribution to the free energy of binding from the covalent energy.
c Contribution to the free energy of binding from the lipophilic energy.
d Contribution to the free energy of binding from the electrostatic solvation energy.
e Contribution to the free energy of binding from the van der Waals energy.
f DG Bind Energy.
In this study various flavonoids were docked against selected proteins. Following this, MM-GB/SA was used as a post docking process to evaluate the validity of the docking results. All energy values obtained from the MM-GB/SA calculation are given in Table 4. Following analysis of the computed energies, it is apparent that nonpolar forces as well as van der Waals forces comprise the main energetic contributions which stabilize protein-flavonoid interactions and bonds. Conversely, Columbic and polar solvation energy contributions have an inverse effect on bond formation and complex stabilization. Typically, results of the MMGBSA method (free energy of binding, ΔGbind) are in the range of -37.403 kcal/ mol to 0.001 kcal/ mol. The respective values computed by this method for selected compounds 28 ,27, and 21 are -30.04 kcal/ mol, -23.608 kcal/ mol and -30.04 kcal/ mol, close to experimental maximum values. Thus, these finding support the docking results. Compound 13 which had the best bioactivity of the compounds studied shows a value of -26.864 kcal/ mol, ΔGbind, indicating an average relationship. The van der Waals forces were found to be best for compounds 4 and 17, and were -39.25 kcal/ mol and -35. 21 kcal/ mol respectively. For the selected compounds these values were -30.37 kcal/ mol, -31.73 kcal/ mol and -31.43 kcal/ mol for compounds 28, 27, and 21, respectively. Again, these are very close to the maximum value which may be obtained. As such, these results are also in support of docking results and show a strong correlation with experimental data. If polar solvation energy becomes positive, it serves as a signal of good results and also shows exothermic reaction nature. According to data obtained, compound 9 presented the best ΔGsolvGB value, with a value of +54.25 kcal/ mol. For the selected compounds these values were 38 kcal/ mol, 41.36 kcal/ mol and 38.67 kcal/ mol for compounds 28, 27, and 21, respectively. Electrostatic forces (nonpolar solvation energy [ΔGSA]) serve as the driving force behind ligand-receptor binding, and according to the results compound 4 and 9 were the best, showing values of -11.620 kcal/ mol and -10.833 kcal/ mol respectively. Values obtained for selected compounds were -7.24 kcal/ mol, -7.44 kcal/ mol and 8.00 kcal/ mol for compounds 28, 27, and 21, respectively. The overall results were found to validate the experimental data.
Conclusion
In this study, to find out potential anti-urease analogues, computational studies were performed with the series of Flavonoids derivatives. So that, it can be learned that how targeted ligands produce inhibitory activity having bound with receptors. The accuracy of prediction obtained after the comparison between different docking protocols; consequently, good affinity with receptor observed. Hydrogen bond interactions found from the results of docking and simulation, which create the bond with critical amino acid residues and Gln364, Asn168, Ala365, Glu222, Leu318, Arg338, Glu311, Gly279, Thr251 and Ala275 play the vital role. Comparative study of RMSF and RMSD shows that structures of docked ligands were more stable than original structures. It revealed that electron donating groups and less bulky substituent attached to ligand increase the electron density itself, which can affect the binding ability of amino acids and ligands, eventually, make these inhibitors potent anti-urease therapeutic agent.
Acknowledgments
From the Devi Ahilya University, we thank Pradeep Gupta for simulation studies and computation support.
References
- Middleton, E. J. Adv. in Exp. Med. and Bio. 1998, 439,175.
- The Gold Book, 2009.
- Koes, R.; Verweij, W. Quattrocchio, F. Tre. In Pla. Sci. 2005, 10, 236.
- Mol, J.; Grotewold, E.; Koes, R. Tre. Pla.Sci. 1998, 3, 212.
CrossRef - Winkel-Shirley, B. Curr. Opin. Plant Biol. 2002, 5, 218.
CrossRef - Bradshaw, H. D.Schemske, D. W. Nat. 2003, 426, 176.
CrossRef - Field, T. S.; Lee, D. W.; Holbrook, N. M. Plant Physiol. 2001, 127, 566.
CrossRef - Pollastri, S.; Tattini, M. Ann. Bot. 2011, 108, 1225.
CrossRef - Ferrer, J.; Austin, M.; Stewart, C. J.; Noel J. Plant Physiol. Biochem. 2008, 46, 356.
CrossRef - Manach, C.; Scalbert, A.; Morand, C. Am J. Clin. Nutr. 2004, 79, 727.
CrossRef - Iwashina, T. Bull. Natl. Mus. Nat. Sci. 2013, 39, 25.
- Panche, A. N.; Diwan, A. D.; Chandra, S. R. J Nutr Sci. 2016; 5, e47.
CrossRef - David, A. V. A.; Arulmoli, R. Pharmacogn Rev. 2016, 10, 84.
CrossRef - http://www.ars.usda.gov/nutrientdata/flav.
- Sangeetha, K. S. S.; Umamaheswari, S. Int. J. Pharm. Sci. Res. 2016; 7, 3924.
- Tiwari, S. C.; Husain, N. Indian J.Sci.Res. 2017, 12, 193.
- Xiao, Z.P.; Wang, X.D. J. Agric. Food Chem., 2012, 60, 10572.
CrossRef - Schrödinger Release 2017-1: LigPrep, Schrödinger, LLC, New York, NY, 2017.
- Hayes, M. J.; Stein, M.; Weiser, J. J.Phys.Chem.A. 2004, 108, 3572.
CrossRef - Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G. Nuc. Aci. Res., 2000, 28, 235.
CrossRef - Prime, Tool for energy calculations, Schrödinger, LLC, New York, 2017.
- Glide, Tool for docking, Schrödinger, LLC, New York, NY, 2017.
- Schreiter, E. R.; Sintchak, M. D. Nat.Struct.Biol. 2003, 10, 794.
CrossRef - Pearson, M. A.; Michel, L. O. Biochem. 1997, 36, 8164.
CrossRef - Desmond Molecular Dynamics System, Maestro-Desmond Interoperability Tools, D. E. Shaw Research, New York, NY, 2017.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


