Stability Indicating UHPLC Method Development and Validation for Estimation of Eltrombopag and its Related Impurities in Tablet Dosage form
Tharlapu Satya Sankarsana Jagan Mohan1,2 , Khagga Mukkanti4 and Hitesh A Jogia3
, Khagga Mukkanti4 and Hitesh A Jogia3
1Research and Development, Jawaharlal Nehru Technological University, Kakinada-533003, India.
2Epione Labs Private LTD, Nacharam Industrial Area, Hyderabad-500076, India.
3Janssen Pharmaceuticals, Jogeshwari (E), Mumbai-400060, India.
4Institute of Science and Technology, Jawaharlal Nehru Technological University, Kukatpally, Hyderabad-500085, India.
Corresponding Author E-mail: victoryjagan1@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/340262
Article Received on : January 01, 2018
Article Accepted on : March 05, 2018
A novel, agile, mass compatible, stability indicating isocratic method developed by using ultra high performance liquid chromatographic (UHPLC) for estimation of Eltrombopag along with its impurities in tablet formulation. Achieved a good separation on Agilent SB C8 (50× 3.0 mm, 1.8 μ) column and mobile phase composed of acetonitrile and 0.1% glacial acetic acid buffer in the ratio of 60:40 v/v at 0.4 ml/minute flow rate and 25°C column temperature within runtime of 15 minutes. Eltrombopag was detected at 230 nm. The proposed method proves its stability indicating power by resolving Eltrombopag peak from its process impurities and degradation products with more than resolution 2.0. The proposed method is not only simple but capable to separate its potential degradant. The intended method is efficient to determine Assay of Eltrombopag in the existence of its impurities within the same run. The retention time observed for Eltrombopag is about 3.9 minutes. The designed method is validated for precision, accuracy, sensitivity, robustness and specificity parametersaccording to ICH guidelines. Eltrombopag was exposed to stress conditions in acidic, basic, thermal, water, oxidative, humidity and photo degradation. Noticeable degradation is observed in oxidative hydrolysis.
KEYWORDS:Eltrombopag; Validation; UHPLC; Stability Indicating Method; Forced Degradation; Methanol
Download this article as:| Copy the following to cite this article: Mohan T. S. S. J, Mukkanti K, Jogia H. A. Stability Indicating UHPLC Method Development and Validation for Estimation of Eltrombopag and its Related Impurities in Tablet Dosage form. Orient J Chem 2018;34(2). |
| Copy the following to cite this URL: Mohan T. S. S. J, Mukkanti K, Jogia H. A. Stability Indicating UHPLC Method Development and Validation for Estimation of Eltrombopag and its Related Impurities in Tablet Dosage form. Orient J Chem 2018;34(2). Available from: http://www.orientjchem.org/?p=44713 |
Introduction
Eltrombopag is an approved drug for adult patients suffering from chronic immune thrombocytopenicpurpura (ITP). The symptoms are insufficient response to immune globulins, or splenectomy,corticosteroidsand having oral bioavailability. Use of Eltrombopag in lower dose is being essential to shorten the bleeding risk by increasing the platelet count and the dose can be adjusted upon the platelet count response. It is also used for thrombocytopenia treatment in patients withhepatitis C virus and chronic liver disease (1-3). Chemically Eltrombopag Olamine is 3′-{(2Z)-2-[1-(3,4-dimethylphenyl)-3-methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene]hydrazino}-2′-hydroxy-3-biphenylcarboxylic acid – 2-aminoethanol (1:2)(Figure 1) and the molecular weight of Eltrombopag Olamine is 564.65 and 442.5 for Eltrombopag (4). Few analytical methods reported in the literature for the content estimation of Eltrombopag in drug substance, biological samples using high performance liquid chromatography (HPLC) and Mass Spectroscopy (MS) (5-7). There is one more reported method for the Eltrombopag quantification in pharmaceutical bulk and formulations by using ultra performance liquid chromatography (UPLC) (8). The purpose of this research was to develop an assay method which is stability indicating for estimation of Eltrombopag in a pharmaceutical formulations by using ultra high performance liquid chromatography (UHPLC) and separating its degradants along with process related impurities. To the simplest of our information no methodology is accessible in the literature by separating the impurities, 1, 2, 3 (Figure 1) using only 15 minutes runtime.
The objective of stability indicating method is to resolve all the major degradants and potential process related impurities by achieving a proper mass balance during the forced degradation. Stability indicating method is capable to measure the active substance and all its degradation products unequivocally in presence of excipients present in the respective formulation (9). The power of any stability indicating analytical method is to be proved in stress study by identifying its degradation products which in turn establishes the innate stability of the molecule and its degradation pathways (10).
The key objective of the current research was to develop a validated reverse phase UHPLC method which is stability indicating for the quantitative estimation of Eltrombopag and its process impurities and possible degradants during forced degradation. In the current study Eltrombopag, three of its process related impurities are: Impurity-1 (Figure 1b), Impurity-2 (Figure 1c) and Impurity-3 (Figure 1d).
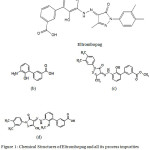 |
Figure 1: Chemical Structures of Eltrombopag and all its process impurities (a) Eltrombopag (b) Imp-1 (c) Imp-2 (d) Imp-3
|
Materials and Methods
Chemicals and reagents
The Eltrombopag active pharmaceutical ingredient and three process related impurities (Impurity-1, Impurity-2 and Impurity-3) and the finished product tabletswas gifted by Epione Labs (Nacharam, Hyderabad, India).Acetonitrile, glacial acetic acid, hydrochloric acid and hydrogen peroxide, sodium hydroxide were procured from Merck, Germany. Ultra pure water used in this study iscollectedfrom Milli-Q plus purification system.
Equipment
AnUHPLC system (Make: Shimadzu, Kyoto, Japan)armed with a diode array detector was used. The output signal was processed using Lab solutions software. Samples for thermal stability study wereexposed to heat in an oven (Make: Thermolab, India).
Chromatographic conditions
Final separation was obtained onAgilent SB C8 50× 3.0 mm, 1.8 μ column with mobile phase composed of acetonitrile and 0.1% glacial acetic acid buffer (1.0 ml of glacial acetic acid in 1 L of water) in 60:40 v/v ratio and filtered with 0.45µmmembrane filter. The mobile phase flow rate was kept at 0.4 mL/min by maintaining the column temperature at 25°C, monitored at 230 nm detector wavelength. Samples were injectedfor a run time of about 15 minutes with an injection volume of2µL.Used methanol as diluent. Calculations were performed by using externalstandardization methodology.
Standard solution
Eltrombopag 1000 µg/ml standard stock solution is prepared by dissolving the equivalent amount in diluent.
Further a 0.5 µg/ml ofEltrombopag standard solution of was prepared in further dilutions using diluent.
System suitability solution
Eltrombopag 1000 µg/ml and all impurities 0.5 µg/mlare prepared by dissolving equivalent amount of API and spiking impurities in diluent.
Sample Solution
Crush 20 tablets to a fine powder and transferred 100 mg of powder equivalentof Eltrombopag in a100 mL volumetric flask. Diluent of 60 mL was added to it and sonicated for 30 min with and make up to volume with diluent. Filter the solution using 0.45µmfilter.
A spiked sample solution is prepared by spiking all three known impurities at the concentration of 0.5% with respect to sample concentration of 1000 µg/ml.
Method development and Optimization of the chromatographic conditions
The main objective of the research is to develop a stability indicating method for the separation Eltrombopag and its related compounds by using UHPLC. Several trails were conducted during the stability indicating method development using UHPLC for the analysis of Eltrombopag and their related compounds in pharmaceutical tablet formulation. Tried with various compositions of mobile phase which consists of buffer with solvents like acetonitrile and methanol, but the combination of buffer and acetonitrile appeared in a lack of symmetry for Eltrombopag and their related compound peaks. The finest results were obtained in the mobile phase consist of acetonitrile and 0.1% glacial acetic acid buffer (1.0 ml of glacial acetic acid in 1 L of water) in 60:40 v/v rationat flow rate of 0.4 mL/min, using Agilent SB C8 50× 3.0 mm, 1.8 μ column,column temperature of 25°C using DAD detector at 230 nm, and able to resolve Eltrombopag and its three impurities (Imp-1 to Imp-3) (Fig. 2) by meeting the system suitability requirement with a resolution of more than 2.0 between all adjacent peaks. Evaluated the response factor for the impurities related to Eltrombopag (Table I). Using the above chromatographic conditions achieved a good separation with a total runtime of 15 minutes.
Individual solutions of Eltrombopag and its process impurities were scanned in the range of 200 nm to 400 nm using a diode array detector. Found that all the compounds are showing maximum absorbance at about 230 nm and hence 230 nm was opted for method detection.
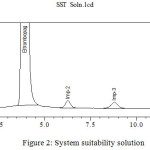 |
Figure 2: System suitability solution |
Table 1: System suitability data.
| Name | RT (min) | RRTa | Resolutionb | Tailing Factor | RRFc |
| Imp-1 | 1.081 | 0.28 | – | 1.3 | 2.11 |
| Eltrombopag | 3.852 | 1.00 | 11.4 | 1.1 | 1.00 |
| Imp-2 | 6.103 | 1.58 | 5.9 | 1.1 | 0.83 |
| Imp-3 | 8.519 | 2.21 | 4.8 | 1.2 | 0.90 |
Relative retention times (RRT) were calculated against the retention time (RT) of Eltrombopag.
Resolution was calculated between two adjacent peaks.
Relative response factor (RRF) was computed against Eltrombopag peak response.
Results
Method validation
Specificity and forced degradation studies
Specificity shows the ability of the method to quantify the response of the analyte in accompany of its degradation products and free from blank and placebo interference. Likely degradation product will be identified in stress degradation studies indicates the stability indicating nature of the method. Stress degradation studies for Eltrombopag tablets were executed under acidic hydrolysis (For 60 min in 0.1M HCl at 60°C), basic hydrolysis (For 60 minat 60°C in 0.1 M NaOH), hydrolytic oxidation (For 60 min in hydrogen peroxide (10%) at 60°C), and thermal condition (For 12 hrs at 105°C),Refluxed at 60°C with water for about 60 minutes.Exposed the samples to humidity study at 25°C/ 90 % RH about 15 days and exposed the sample to Light at 200 Watt-hours. The samples prepared in the above conditions were filtered with 0.45 µm filter before dilution. Water bath (with temperature controller) was used to carry out the stress degradation studies.
Eltrombopag shows a significant degradation in peroxide oxidation (Fig. 3b). Eltrombopag was degraded into Imp-1, Imp-2 and unknown impurity at RRT about 1.29 by oxidative hydrolysis. The mass balance found to be more than 95% for the degraded samples in all the stress conditions (Table II). Verified peak purity for everystress sample solution using HPLC-PDA system and the results found to be passed as per the software acceptance criteria, which demonstrates analyte peak homogeneity (Free from overlapping of peaks).
 |
Figure 3: Eltrombopag forced degradation chromatograms (a) Base hydrolysis (b) Peroxide oxidation |
Table 2: Stress degradation data of Eltrombopag
| Stress Condition index | % Degradation | Mass Balance | Peak purity |
| Acid (For 60 min in 0.1M HCl at 60°C) | 0.04 | 99.6 | 1.000 |
| Base (For 60 min in 0.1 M NaOH at 60°C) | 0.14 | 98.7 | 1.000 |
| Peroxide (For 60 min in H2O2(10%) at 60°C) 2.23 98.6 1.000 | |||
| Water (at 60°C, 60 min) | 0.05 | 99.5 | 1.000 |
| Thermal (For 12 hrs at 105°C) | 0.05 | 99.5 | 1.000 |
| Humidity (For 15 Days at | |||
| 25°C and 90 % RH) | 0.05 | 100.0 | 1.000 |
| Light (200 Watt/Hrs) | 0.04 | 98.8 | 1.000 |
aMass Balance= % Degradation + % Assay
Precision
Precisionstudy for the intended method was assessed by injecting Eltrombopag sample along with 0.5%level of its three impurities (0.5% of impurity concentration with respectto 1000 mg/mL Eltrombopag) on six individual samples. The percent RSD of spiked Impurities 1, 2, and 3are less than 5%, proves good method precision (Table III).
Table 3: Regression and precision data for Eltrombopag.
| Compound | LOD (%) | LOQ (%) | Slope (b) | Intercept (a) Precision | Precision (%RSD) | LOQ (%RSD) |
| Imp-1 | 0.001 | 0.010 | 404897 | -9088 | 1.0 | 7.0 |
| Eltrombopag | 0.018 | 0.020 | 191735 | 2850 | 0.9 | 4.4 |
| Imp-2 | 0.013 | 0.038 | 158891 | 1500 | 0.6 6.5 | |
| Imp-3 | 0.012 | 0.035 | 172570 | -303 | 0.8 7.0 |
Linearity
Linearity of the method was established by preparing a series of six concentrations (i.e., LOQ, 10, 20, 40, 100, and 200%) with 1.0% thought of to be 100% by linear least square analysis method. Correlation coefficient (r), slope and y-interceptfor the three impurities are presented in Table III. The obtained correlation coefficientis more than 0.999, for all impurities indicating a linear response of the method.
Limit of Quantification (LOQ) and Limit of Detection (LOD)
The LOQ and the LOD for all the threeprocess impurities of Eltrombopagwere assessed at the concentrations where the signal-to-noise ratios detected were 3:1 ratio and 10:1 ratio, respectively. LOQ level precision was also determined by analysing six individual consecutive preparations of the threeprocess impurities by calculating the RSD (%) (Table III).
Accuracy
For all the three impurities the percentage recovery in triplicate was determined for all the three impurities at 0.5, 1.0, and 1.5% levels with respect the Eltrombopag concentration (1000 mg/mL) (Table IV).
Table 4: Accuracy (n=3) data for Eltrombopag.
| Level | Imp-1 (%RSD) | Imp-2 (%RSD) | Imp-3 (%RSD) | Eltrombopag (%RSD) |
| 50% | 104.8(1.6) | 100.3(0.5) | 105.0(2.4) | 101.5(0.9) |
| 100% | 106.6(0.6) | 102.3(1.2) | 100.9(1.5) | 100.8(0.9) |
| 150% | 103.7(3.3) | 100.4(4.0) | 98.1(3.0) | 102.1(0.6) |
Robustness
Robustness determines the capability of the method to remain unchanged by small planned variations in the method parameters. The conditions studied were flow rate(altered by ±0.05 mL/min), column temperature (alteredby ±5°C) and organic phase variation (alteredby ±10%).Resolution between adjacent peaks is more than 2.0 in all the conditions (Table V).
Table 5: Robustness data.
| Resolutiona | 0.35ml/minACN | 0.45ml/min | 20°C | 30°C110% | 90% ACN |
| Imp-1 | – | – | — | — | |
| Eltrombopag | 10.0 | 10.8 | 11.3 | 11.47.9 | 10.0 |
| Imp-2 | 4.5 | 5.4 | 5.34.2 | 5.45.4 | |
| Imp-3 | 3.4 | 4.2 | 4.03.0 | 4.24.2 |
aResolution between adjacent peaks was calculated.
Solution stability
Standard and test solutions stability were verified by keeping them on benchtop for 48 hours. Both the solutions are found to be stable at benchtop and no significant change was observed for all the three impurities.
Conclusion
An easy, specific, reliable and mass compatible isocratic method usingUHPLC-DAD wasdeveloped for the quantitative estimation of Eltrombopag in tablet formulation. This was the first method for separation and quantification of Eltrombopag and its process related impurities in comparison with reported methodologies. The current method was successfully resolvedall the degradants, three known impurities and estimate the active component perfectly. Validated the developed methodaccording to ICH guidelines and found to bespecific,linear, accurate, robust, precise, and rugged.Proposed chromatographic method with a short run time of 15 minutes permits the analysis of a many samples in a day. The developed methodology has proved the stability-indicating power, and hence it can be used for regular analysis throughout stability studies in quality control laboratories.
Acknowledgments
The authors are grateful to themanagementof Epione Labs Pvt Ltd for their support.Authorswishtoacknowledgethe researchgroupforprovidingthesamplesforourresearch.
References
- Deng, Y.; Madatian, A.; Wire, M.B.; Bowen, C.; Park,J.W.; Williams,D.; Peng,B.; Schubert,E.; Gorycki,F.; Levy,M.; Gorycki,P.D. Metabolism and disposition ofeltrombopag, an oral, nonpeptide thrombopoietin receptor agonist, in healthy human subjects,Drug Metab Dispos, 2011,39(9), 1734–1746.
CrossRef - Serebruany, V.L.; Eisert,C.; Sabaeva,E.; Makarov,L. Eltrombopag (Promacta), athrombopoietin receptor agonist for the treatment of thrombocytopenia: current and future considerations, Am. J. Ther,2010, 17(1), 68–74.
CrossRef - Allred, A.J.; Bowen, C.J.; Park, J.W.; Peng, B.; Williams, D.D.; Wire, M.B.; Lee, E. Eltrombopag increases plasma rosuvastatin exposure in healthy volunteers, Br. J. Clin. Pharmacol, 2011, 72(2), 321-329. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/promacta.pdf(accessed Jan1, 2018).
CrossRef - Wire, M.B.; Mclean, H.B.; Pendry, C.; Theodore, D.; Park, J.W.; Peng, B. Assessment of the pharmacokinetic interaction between eltrombopag andlopinavir–ritonavir in healthy adult subjects, Antimicrob. Agents Chemother, 2012, 56(6), 2846–2851.
CrossRef - Williams, D.D.; Peng, B.; Bailey, C.K.; Wire, M.B.; Deng, Y.; Park, J.W.; Collins, D.A.; Kapsi, S.G.; Jenkins, J.M. Effects of food and antacids on the pharmacokinetics of eltrombopag in healthy adult subjects: two single-dose,open-label, randomized-sequence, crossover studies,Clin. Ther, 2009, 31(4),764–776.
CrossRef - Rambabu, M.; Ramakrishna, G.; Nageswara, R.P.; Sridhar, S.; Srinubabu, M.; Ajitha, M. Liquid chromatography–tandem mass spectrometric assay for eltrombopag in 50 µL of human plasma: A pharmacokinetic study,J. of Pharm. and Biomed. Ana, 2014, 98,68–73.
CrossRef - Mahaboob, B.D.; Venkata, R.G.; Shobha, R.T.; Susithra, E.; Rajasekhar, C. RP-UPLC-MS assay method for eltrombopag: Application in pharmaceuticals, human plasma and urine samples,Asian J. Chem, 2015, 27(12),4615–4619.
CrossRef - Monika, B.; Saranjit, S. Development of validated stability indicating assay methods-critical review; J. of Pharm. and Biomed. Ana, 2002, 28, 1011-1040.
CrossRef - ICH harmonized tripartite guideline, stability testing of new drug substances and products, Q1A (R2),2003.
- ICH of technical requirements for the registration of pharmaceutical for the human use, validation of analytical procedures: text and methodology, ICH, Q2 (R1), 2005.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


