Eco-friendly Access to β-ketoamides: One-step Catalyst-and Solvent-free Amidation of β-ketoesters Under Microwave Irradiation
Khadidja Dechira1,2 , Assya Taleb1, Aouicha Benmaati1, Salih Hacini1 and Hadjira Habib Zahmani1
, Assya Taleb1, Aouicha Benmaati1, Salih Hacini1 and Hadjira Habib Zahmani1
1Laboratory of Fine Chemistry, Faculty of Exact and Applied Sciences, University of Oran1 Ahmed Benbella, B.P -1524 Menouar, 31000 Oran, Algeria.
2Scientific and Technical Research Center for Physico-Chemical Analysis (C.R.A.P.C.) B.P 384, Bou-Ismail RP 42004 Tipaza, Algeria.
Corresponding Author E-mail : habibzahmanihadjira@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/340117
A highly efficient and facile catalyst- and solvent-free one step amidation of β-ketoesters, without using any additional reagents, is described. Therefore, β-ketoamides are obtained in good to excellent yields by condensation of β-ketoesters with various primary or secondary amines. This eco-friendly protocol has been developed under microwave irradiation.
KEYWORDS:Amidation; β-ketoesters; β-ketoamides; catalyst-free; solvent-free; Microwave irradiation
Download this article as:| Copy the following to cite this article: Dechira K, Taleb A, Benmaati A, Hacini S, Zahmani H. H. Eco-friendly Access to β-ketoamides: One-step Catalyst-and Solvent-free Amidation of β-ketoesters Under Microwave Irradiation. Orient J Chem 2018;34(1). |
| Copy the following to cite this URL: Dechira K, Taleb A, Benmaati A, Hacini S, Zahmani H. H. Eco-friendly Access to β-ketoamides: One-step Catalyst-and Solvent-free Amidation of β-ketoesters Under Microwave Irradiation. Orient J Chem 2018;34(1). Available from: http://www.orientjchem.org/?p=42507 |
Introduction
Catalyst-free methodologies for synthesis of organic compounds are of great interest for the development of a sustainable chemistry. In addition to the simplicity of the procedure and the reduced cost, this also makes it possible to considerably reduce the chemical waste in our environment [1]. In this communication, we report a simple and rapid, catalyst-free one-step synthesis of various β-ketoamides. These products are very useful compounds to access to a wide variety of Chemical structures (heterocycles, natural products) and targets of pharmaceutical research [2]. Various synthetic methods have been reported in the literature to prepare amides, but the processes of access to β-ketoamides are rather limited. Among these, mention may be made of the nucleophilic amidation of electrophiles such as β-ketoacids [3], β-ketoesters [4], β-thioesters [5] and keten dimmers [6]. Other methods are based on Dieckmann cyclization of amidoesters [7], treatment of carboxamides with phosgene or phosphoryl chloride [8], diketenes [9], enzymatic hydrolysis of β-ketonitriles [10] and aminolysis of β-ketoesters [11], organocatalytic multicomponent of polyfunctional substrates [12]. β-Ketoamides can also be prepared via Wolf rearrangement of diazodiketones in the presence of amines [13].
Among these approaches, the amidation is the most frequently used reactions. In general this process involves specific reaction conditions as the use of a specific catalysts, vigorous reaction conditions and prolonged reaction times. These reactions often lead to unsatisfactory yields of 1,3-ketoamides due to the competitive enaminoester formation, or requires uncommercial starting materials. In our efforts to develop greener and simpler amidation method, we focused on the catalyst-free approach. Therefore, a facile one-pot process for the synthesis of N-mono- and di-substitued β-ketoamides has been elaborated from readily available cyclic β-ketoesters in the presence of amines. Amines, amongst the most important reagents in organic synthesis by their basic and nucleophilic characters, are used in our strategy as reagent and as catalyst. Realizing a combination process of temperature and pressure to activate this reaction was a key contribution.
Materials and Methods
Solvents and reagents were purchased from Sigma Aldrich and used as received. The reactions under microwave irradiation were performed in a microwave apparatus Anton Paar, Monowave 300 model power 850 W. The reactions were monitored by TLC visualized by UV (254 nm) and/or with p-anisaldehyde. The melting points were determined on an M-560 melting point apparatus and are uncorrected. Purification of reaction products was carried out by flash chromatography using silical-gel (40-63 mm) eluted with AcOEt/PE or EtO2/PE. NMR data were recorded at 300 or 400 MHz (Bruker Avance spectrometers) in CDCl3 using as internal standards the residual CHCl3 signal for 1H NMR (d = 7.26 ppm) and the deuterated solvent signal for 13C NMR (d = 77.16ppm). IR spectra were obtained with a Alpha-p Brucker ATR diamond FT-IR spectrophotometer.
Results and Discussion
The environment-friendly methods developed in our work have now evolved to microwave-accelerated solvent free aminolysis procedure. In this context, to appraise the combination of temperature and pressure effect we tested the feasibility of amidation at several temperatures. We initiated our studies of the amidation of cylohexanone-ester 1a with morpholine 1b. For the experimental procedure we simply mixed 1equiv. of ester 1a with 1,1 equiv of amine 1b and irradiated the reaction mixtures in a MW oven in the absence of solvent (Schema 1). As summarized in Table 1, formation of β-ketoamide 1c generally required high temperatures; at 80°C, 100°C and 120°C the isolated yields of amide just slightly increased. On the other hand, we observed that an increased temperature from 160°C to 180°C did increase the conversion from 94% to quantitative and speed up the time of reaction. We show here that the ester 1a and amine 1b can only react when they collide by heating the mixture, as particles move faster and collide more frequently.
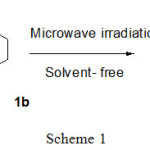 |
Scheme 1 Click here to View scheme |
Table 1: Reaction temperatures tested for the amidation of 1a
|
Entry |
Temp. (°C) |
Time (min) |
yield % (1c) |
|
1 |
80 |
120 |
Trace |
|
2 |
100 |
120 |
20 |
|
3 |
120 |
120 |
37 |
|
4 |
160 |
60 |
95 |
|
5 |
180 |
30 |
Quantitative |
In a second stage, the scope of the reaction was investigated with variety of secondary amines 1b-6b and cyclic β-ketoesters (five (C5) and six (C6) membered rings). The solvent-free methodology appeared to be applicable to prepare a wide range of N-disubstitued β-ketoamides compounds, as reported Table 2.
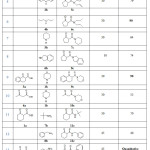 |
Table 2: Solvent-free and catalyst-free synthesis of various β-ketoamide under MW irradiation. Click here to View table |
A variety of secondary amines were efficiently converted to the corresponding products in moderate to excellent yields (52%-quantitative). The reaction of cyclopentanone-ester 2a with morpholine and dipropylamine (Table 2, entries 4 and 6) provide excellent yields of 4c and 6c. Actually, in most of the cases, aminolysis occurred to be more efficient under solvent-free conditions (table 2, entries 1, 4, 6, 7 and 8). Furthermore we studied the reactivity of morpholine toward ethyl 1-oxo-1,2,3,4-tetrahydronaphtalene-2-carboxylate 3a and acyclic ketoester; ethyl acetoacetate 4a (Table 2, entries 9 and 10). Aminolysis of 3a gave the corresponding amide 9c with good yield (90%), however, only moderate catalytic activity was observed in the case of ethyl acetoacetate 4a which product yield 10c do not exceed 52%.
Following the same protocol, we directed our attention to the amidation of primary amine which are known by their low reactivity. All reactions were performed using 1.1 equiv of amine to form N-disubstitued ketoamide from Ketoesters and were obtained with moderate to good yield (40% -quantitative). As is evident from Table 2, aniline and tertbutylamine produced the best yields compared to other amines (Table 2, entries 13, 14 and 18). Benzylamine and furfurylamine, the less reactive amines, produce slightly low yields (40 and 50%), (Table 2, entries 12 and 15). In evaluating the amine catalysts effects we noticed that the aminolysis performance depends strongly on the amines basicity as shown in Table 3. It is clear that the yield of the reaction decreased with increasing pKa.
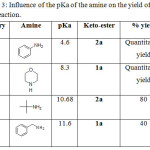 |
Table 3: Influence of the pKa of the amine on the yield of the reaction. Click here to View table |
Thus, this new approach allows us to understand the basic catalytic activity of amine in aminolysis process. In this method, reactions are faster, safer and with higher chemical yields, that is why this method becomes superior to the conventional method. Furthermore these results reveal that the present method is useful for aromatic as well as for aliphatic amines.
Experimental
General procedure for the synthesis of β-Ketoamides
To an open glass tube (G4), were added β-ketoesters (1.0 mmol) and primary or secondary amines (1.1 mmol) in solvent – free. The tube was positioned in the centre of the microwave cavity, and irradiated (850 W) until the temperature reached 180°C for 15 a 60 min. Purification of the resultant product by short column chromatography over silica gel (40–63 mesh) to obtain the pure products.
2-(morpholine-4-carbonyl)cyclohexanone (1c)
Brown solid; mp = 77.7 °C; Rf (EtO2/PE: 4/6) = 0.25; IR: ν 2941, 2859, 1705, 1626, 1186, 1109 cm-1; 1H NMR (300 MHz, CDCl3): δ = 3.82 (dt, J= 13.60, 4.34 Hz, 1H) , 3,71 (t, J= 4.81 Hz, 2H) , 3,67 (t, J= 4.81 Hz, 2H), 3.57 – 3.47 (m, 2H), 3.34 (dd, J= 6.04, 3.96 Hz, 1H), 2.54(dt, J= 6.04, 3.96, 1H), 2.41 – 2.17 (m, 2H), 2.15 – 1.96 (m, 3H), 1.92 – 1.81 (m, 1H), 1.79 – 1.62 (m, 2H) ppm; 13C NMR (75 MHz, CDCl3):δ = 207.35(C), 168.06(C), 66.82(CH2), 66.59(CH2), 54.12(C), 46.26(CH2), 42.29 (CH2), 41.98 (CH2), 30.17(CH2), 27.09(CH2), 23.64(CH2) ppm.
N,N-diallyl-2-oxocyclohexanecarboxamide (2c)
Brown oil; Rf (EtO2/PE: 6/4) = 0.33; IR: ν 3082, 2983, 1720, 1631, 1190, 924 cm-1; 1H NMR (300 MHz, CDCl3): δ = 5.80 – 5.69 (m, 2H), 5.25 – 5.08 (m, 4H), 4.28 (dd, J = 15.5, 4.3 Hz, 1H), 3.84 – 3.79 (m, 1H), 3.72 (dd, J = 5.0, 1.6 Hz, 1H), 3.68 (d, J = 5.5 Hz, 1H), 3.48 (dd, J = 10.0, 5.3 Hz, 1H), 2.53 (dt, J = 13.3, 4.5 Hz, 1H), 2.33 – 2.14 (m, 2H), 1.99 (dqd, J = 12.4, 5.2, 2.7 Hz, 3H), 1.85 – 1.73 (m, 1H), 1.68 – 1.57 (m, 1H) ppm; 13C NMR (75 MHz, CDCl3): δ = 207.60(C), 169.95(C), 133.71(CH), 133.08(CH), 117.07(CH2), 116.67(CH2), 54.65(CH), 49.50(CH2), 48.17(CH2), 42.17(CH2), 30.69(CH2), 27.24(CH2), 23.85(CH2) ppm.
2-(4-phenylpiperazine-1-carbonyl) cyclohexanone (3c)
White solid; mp = 102.9°C; Rf (EtO2/PE: 6/4) = 0.28; IR: ν 2861, 1692, 1640, 1596, 1183, 729, 669 cm-1; 1H NMR (300 MHz, CDCl3): δ = 7.34 (t, J = 7.6 Hz, 2H), 7.25 – 6.93 (m, 3H), 4.17 – 3.97 (m, 1H), 3.83 – 3.75 (m, 1H), 3.58 (ddd, J = 21.9, 10.9, 9.9 Hz, 2H), 3.26(s, 3H), 2.63 – 2.55 (m, 1H), 2.45 – 2.35 (m, 1H), 2.30 – 2.22 (m, 1H), 2.17 – 1.94 (m, 3H), 1.92–1.66 (m, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ = 207.43(C), 167.97(C), 150.71(C), 129.25(CH), 120.69(CH), 116.75(CH), 54.26(CH2), 49.67(CH2), 45.67(CH2), 41.98(CH2), 41.79(CH2), 30.94(CH2), 30.29(CH2), 27.13(CH2), 23.60(CH2) ppm.
2-(morpholine-4-carbonyl)cyclopentanone (4c)
White oil; Rf (EtO2/PE: 6/4) = 0.32; IR: ν 2967, 1735, 1623, 1109, 1067 cm-1; 1H NMR (300 MHz, CDCl3): δ =3.86 (dd, J = 13.4, 1.8 Hz, 1H), 3.78 – 3.67 (m, 3H), 3.54 (ddd, J = 11.3, 2.8, 1.4 Hz, 1H), 3.44 – 3.28 (m, 3H), 2.56 – 2.44 (m, 1H), 2.24 (dd, J = 8.4, 6.6 Hz, 2H), 2.18 – 2.07 (m, 2H), 1.90 – 1.76 (m, 1H), 1.28–1.14 (m, 1H) ppm. 13C NMR (75 MHz, CDCl3): δ = 214.28(C), 166.79(C), 67.13(CH2), 66.88(CH2), 51.62(CH), 46.68(CH2), 42.75(CH2), 38.72(CH2), 27.14(CH2), 21.08(CH2) ppm.
N,N-diallyl-2-oxocyclopentanecarboxamide (5c)
Yellow oil; Rf (EtO2/PE: 4/6) = 0.26; IR: ν 3461, 1738, 1632, 1413, 921 cm-1; 1H NMR (300 MHz, CDCl3): δ = 5.86 – 5.64 (m, 2H), 5.14 (dd, J = 13.7, 3.2 Hz, 3H), 5.09 (d, J = 5.8 Hz, 1H), 4.30 (d, J = 3.2 Hz, 1H), 4.25 (d, J = 4.6 Hz, 1H), 3.80 (dd, J = 18.1, 4.5 Hz, 1H), 3.65 (dd, J = 15.5, 5.9 Hz, 1H), 3.35 (t, J = 8.6 Hz, 1H), 2.55 – 2.39 (m, 1H), 2.26 (dd, J = 8.8, 6.3 Hz, 2H), 2.19– 2.07 (m, 2H), 1.88 – 1.72 (m, 1H) ppm; 13C NMR (75 MHz, CDCl3): δ = 214.60(C), 168.95(C), 133.23(CH), 132.68(CH), 116.85(CH2), 116.37(CH2), 51.97(CH), 49.22(CH2), 48.18(CH2), 38.52(CH2), 27.50(CH2), 20.97(CH2) ppm.
2-oxo-N,N-dipropylcyclopentanecarboxamide (6c)
Brawn oil; Rf (EtO2/PE: 6/4) = 0.29; IR: ν 2872, 1739, 1630, 1509, 1103, 733 cm-1; 1H NMR (300 MHz, CDCl3) : δ = δ: 3.55 – 3.41 (m, 2H), 3.34 (t, J = 8.5 Hz, 1H), 3.18 – 3.06 (m, 2H), 2.48 – 2.36 (m, 1H), 2.29 – 2.23 (m, 2H), 2.19 – 2.08 (m, 2H), 1.89 – 1.76 (m, 1H), 1.34 – 1.20 (m, 4H), 0.93 (dd, J = 15.7, 7.4 Hz, 6H) ppm; 13C NMR (75 MHz, CDCl3) : δ = 214.72(C), 168.72(C), 51.59(CH), 47.86(CH2), 46.04(CH2), 38.48(CH2), 31.36(CH2), 29.76(CH2), 27.68(CH2), 21.01(CH2), 20.03(CH2), 20.00(CH2), 13.76(CH3), 13.72(CH3) ppm.
2-(piperidine-1-carbonyl)cyclopentanone (7c)
yellow oil; Rf (EtO2/PE: 6/4) = 0.36; IR: ν 2851, 1739, 1626, 1433, 1217, 751 cm-1; 1H NMR (300 MHz, CDCl3) : δ = 3.58 (dddd, J = 16.7, 14.4, 10.8, 5.5 Hz, 4H), 2.86 (t, J = 7.2 Hz, 1H), 2.54 – 2.41 (m, 1H), 2.34 – 2.28 (m, 1H), 2.23 – 2.10 (m, 2H), 2.0 – 1.80 (m, 2H), 1.72 – 1.55(m, 6H) ppm; 13C NMR (75 MHz, CDCl3): δ = 214.07(C), 166.66(C), 66.86(CH2), 66.60(CH2), 51.37(CH), 46.42(CH2), 42.46(CH2), 38.23 (CH2), 26.91(CH2), 20.82(CH2) ppm.
2-(isoindoline-2-carbonyl)cyclopentanone (8c)
white solid; mp = 91 °C; Rf (EtO2/PE: 4/6) = 0.29; IR: ν 2892, 1731, 1648, 1594, 1196, 725, 704 cm-1; 1H NMR (300 MHz, CDCl3): δ = 8.21 (d, J = 8.1 Hz, 1H), 7.18 (dd, J = 7.1, 5.9 Hz, 2H), 7.03 (t, J = 7.9 Hz, 1H), 4.54 (td, J =10.1, 6.1 Hz, 1H), 4.07 (td, J = 10.1, 7.3 Hz, 1H), 3.48 (t, J = 10.2, 6.6 Hz, 1H), 3.33 – 3.07 (m, 2H), 2.67 – 2.50 (m, 1H), 2.36 (dd, J = 8.2, 2.6 Hz, 1H), 2.33 (s, 1H), 2.31 – 2.18 (m, 2H), 1.99 – 1.83(m, 1H) ppm; 13C NMR (75 MHz, CDCl3): δ = 214.32(C), 166.81(C), 142.83(C), 131.66(C), 127.49(CH), 124.61(CH), 124.09(CH), 117.32(CH), 54.82(CH2), 48.33(CH2), 38.70(CH), 27.94(CH2), 27.04(CH2), 21.04(CH2) ppm.
2-(morpholine-4-carbonyl)-3,4-dihydronaphthalen-1(2H)-one (9c)
White solid; Rf (EtO2/PE: 5/5) = 0.51; 1H NMR (300 MHz, CDCl3): δ = 8.02 (dd, J = 7.8, 1.1 Hz, 1H), 7.55 (td, J = 7.5, 1.4 Hz, 1H), 7.36 (d, J = 7.7 Hz, 1H), 7.30 (d, J = 7.7 Hz, 1H), 3.95 (dt, J = 3.7, 3.7 Hz, 1H), 3.90 – 3.82 (m, 1H), 3.80 (d, J = 4.7 Hz, 1H), 3.77 (t, J = 4.2 Hz, 3H), 3.64 – 3.43 (m, 3H), 3.12 (dt, J = 16.8, 4.5 Hz, 1H), 3.05 (td, J = 11.4, 5.3, 4.5 Hz, 1H), 2.53 (dtd, J = 13.7, 11.5, 4.8 Hz, 1H), 2.31 (dq, J = 13.6, 4.4 Hz, 1H) ppm. 13C NMR (75 MHz, CDCl3): δ = 194.16(C), 168.39(C), 144.10(C), 133.90(C), 132.00(CH), 128.84(CH), 127.63(CH), 126.86(CH), 66.89(CH2), 51.33(CH), 46.72(CH2), 42.47(CH2), 28.40(CH2), 26.36(CH2) ppm.
1-morpholinobutane-1,3-dione (10c)
Yellow oil; Rf (EtO2/PE: 5/5) = 0.33; IR: ν 2922, 1716, 1623, 1110, 1067 cm-1; 1H NMR (300 MHz, CDCl3): δ = 3.65 (dd, J = 8.3, 5.2 Hz, 6H), 3.55 (s, 2H), 3.41 (s, 2H), 2.26 (s, 3H) ppm; 13C NMR (75 MHz, CDCl3): δ 202.19(C), 165.08(C), 66.62(CH2), 66.51(CH2), 49.66(CH2), 46.73(CH2), 42.09(CH2), 30.30(CH3).
N-tert-butyl-2-oxocyclohexanecarboxamide and N-tert-butyl-2-hydroxycyclohex-1-ene carboxamide (11c)
White solid; mp = 98-100°C; Rf (EtO2/PE: 8/2) = 0.7; IR: ν 3336, 1702, 1630, 1424, 914 cm-1; 1H NMR (400 MHz, CDCl3): δ =14.29 (s, 1H), 6.65 (s, 1H), 5.14 (s, 1H), 3.02 (ddd, J = 9.6, 5.6, 1.1 Hz, 1H), 2.41 – 2.34 (m, 1H), 2.16 – 2.10 (m, 1H), 2.00 – 1.98-1.93 (m, 1H), 1.92 – 1.83 (m, 3H), 1.61 – 1.57 (m, 2H), 1.32 (s, 9H), 1.27 (s, 9H) ppm; 13CNMR (101 MHz, CDCl3): δ =210.66(C), 172.58(C), 169.78(C), 167.87(C), 97.34(C), 56.56 (CH), 51.15(C), 51.01(C), 41.97(CH2), 30.98(CH2), 29.29(CH3), 28.89(CH3), 28.63(CH2), 27.10(CH2), 23.85(CH2), 22.96(CH2), 22.58(CH2), 21.93(CH2) ppm.
N-benzyl-2-hydroxycyclohex-1-enecarboxamide and N-benzyl-2-oxocyclohexanecarbox amde (12c)
White oil; Rf (EtO2/PE: 7/3) = 0.7; 1H NMR (300 MHz, CDCl3): δ =14.16 (s, 1H),7.28 (s, 1H), 7.27 – 7.20 (m, 5H), 6.15 (s, 1H), 4.43 – 4.35 (m, 2H), 3.18 – 3.12 (m, 1H), 2.36 – 2.25(m, 1H), 2.20 – 2.19 (m, 2H), 2.09 – 2.07 (m, 1H), 1.92 – 1.88 (m, 2H), 1.65 – 1.60 (m, 2H) ppm; 13C NMR (75 MHz, CDCl3): δ = 208.63(C), 171.43(C), 169.14(C), 168.36(C), 137.47(C), 137.30(C), 127.57(CH), 127.49(CH), 126.50(CH), 126.42(CH), 126.29(CH), 126.16(CH), 96.05(C), 55.28(C), 42.17(CH2), 41.88(CH2), 40.97(CH2), 30.11 (CH2), 28.21(CH2), 26.12 (CH2), 22.99(CH2), 21.50(CH2), 21.46(CH2), 20.83(CH2) ppm.
2-oxo-N-phenylcyclopentanecarboxamide (13c)
White solid; mp = 88-90°C; Rf (EtO2/PE: 5/5) = 0.4; IR: ν 3050, 1951, 1739, 1664, 1533, 1442, 1311, 1146 cm-1; RMN1H (300 MHZ, CDCl3): δ = 8.74 (s, 1H), 7.56 (d, J = 7.7 Hz, 2H), 7.32 (t, J = 7.9 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 3.15 (t, J = 9.4 Hz, 1H), 2.46 – 2.27 (m, 4H), 2.12 – 2.05 (m, 1H), 1.91 – 1.84 (m, 1H) ppm; RMN13C (300 MHZ, CDCl3): δ = 217.06 (C) , 164.59 (C), 137.83 (C) , 129.10 (CH), 124.43 (CH), 119.97 (CH), 54.81(CH) , 39.22 (CH2), 25.82 (CH2), 20.35 (CH2) ppm.
N-tert-butyl-2-oxocyclopentanecarboxamide (14c)
White solid; mp= 96-98°C; Rf (EtO2/ PE: 8/2) = 0.54; IR: ν 3312, 1741, 1663, 1548, 745 cm-1; 1H NMR (300 MHz, CDCl3): δ = 6.57 (s, 1H), 2.91 (t, J = 9.1 Hz, 1H), 2.43 – 2.22 (m, 4H), 2.11 – 1.99 (m, 1H), 1.98 – 1.73 (m, 1H), 1.36 (s, 9H) ppm; 13C NMR (75 MHz, CDCl3): δ = 217.05(C), 165.66(C), 54.82(CH), 51.24(C), 38.95(CH2), 28.73(CH3), 25.75(CH2), 20.28(CH2) ppm.
N-(furan-2-ylmethyl)-2-oxocyclopentanecarboxamide (15c)
Brown solid; Rf (AcOEt/PE: 1/1) = 0.38; IR: ν 3050, 1951, 1739, 1664, 1533, 1442, 1311, 1146 cm-1; 1H NMR (300 MHz, CDCl3): δ = 7.38 (t, J = 1.8 Hz, 1H), 6.34 (dd, J = 3.1, 1.9 Hz, 1H), 6.26 (dd, J = 3.2, 1.7 Hz, 1H), 4.54 (dd, J = 15.5, 5.8 Hz, 1H), 4.43 (dd, J = 15.5, 5.4 Hz, 1H), 3.05 (t, J = 9.4 Hz, 1H), 2.43 – 2.29 (m, 4H), 2.12 – 2.08 (m, 1H), 1.90 – 1.88 (m, 1H) ppm; 13C NMR (75 MHz, CDCl3): δ = 215.9 (C), 166.7 (C), 151.0 (C), 142.1 (CH), 110.1(CH), 106.9 (CH), 54.1 (CH2), 38.6 (CH2), 36.4 (CH2), 25.8 (CH2), 20.3 (CH2) ppm.
N-allyl-2-oxocyclopentanecarboxamide (16c)
White solid; Rf (EtO2/PE: 7/3) = 0.46; 1H NMR (300 MHz, CDCl3): δ = 6.95 (s, (NH)1H), 5.74 (ddt, J = 17.1, 10.5, 5.4 Hz, 1H), 5.13 (ddd, J = 17.2, 3.1, 1.6 Hz, 1H), 5.04 (ddd, J = 10.3, 2.9, 1.4 Hz, 1H), 3.80 (tt, J = 5.6, 1.6 Hz, 2H), 2.95 (t, J = 9.3 Hz, 1H), 2.31 – 2.22 (m, 4H), 2.19 – 2.0 (m, 1H), 1.80 – 1.67 (m, 1H) ppm; 13C NMR (75 MHz, CDCl3): δ = 216.0 (C), 166.9 (C), 133.7 (CH), 115.7 (CH2), 54.2 (CH), 41.4 (CH2), 38.6 (CH2), 25.9 (CH2), 20.1 (CH2) ppm.
N-allyl-1-oxo-1,2,3,4-tetrahydronaphthalene-2-carboxamide (17c)
White solid; Rf (PE/ ACEt2: 8/2) = 0.75; 1H NMR (300 MHz, CDCl3): δ = 7.95 (dd, J = 7.8, 1.0 Hz, 1H), 7.43 (td, J = 7.5, 1.4 Hz, 1H), 7.25-7.22 (m, 1H), 7.18 (d, J = 7.6 Hz, 1H), 5.87 – 5.71 (m, 1H), 5.15 (ddd, J = 17.2, 3.1, 1.6 Hz, 1H), 5.05 (ddd, J = 10.3, 2.8, 1.4 Hz, 1H), 3.94 – 3.83 (m, 2H), 3.35 (t, J = 7.2 Hz, 1H), 3.05 (dt, J = 16.7, 5.3 Hz, 1H), 2.83 (ddd, J = 23.2, 14.7, 7.4 Hz, 1H), 2.38 (td, J = 7.4, 5.3 22Hz, 2H) ppm; 13C NMR (75 MHz, CDCl3): δ = 196.2 (C), 167.7 (C), 144.6 (C), 134.1 (CH), 133.9 (CH), 131, 7 (C), 128.7 (CH), 127.5 (CH), 126.6 (CH), 115.7 (CH2), 52.8 (CH), 42.0 (CH2), 27.8 (CH2), 25.4 (CH2) ppm.
N-tert-butyl-3-oxobutanamide (18c)
White oil; Rf (EtO2/PE: 7/3) = 0.75; IR: ν 3316, 1714, 1646, 1544, 791 cm-1; 1H NMR (400 MHz, CDCl3): δ = 6.66 (s, 1H), 3.31 (s, 2H), 2.24 (s, 3H), 1.34 (s, 9H) ppm; 13C NMR (75 MHz, CDCl3): δ = 204.87(C), 164.49(C), 51.33(CH2), 50.92(C), 30.99(CH3), 28.35(CH3) ppm.
Conclusion
In conclusion, a highly efficient and facile one-step synthesis of N-mono- and di-substitued β-ketoamides has been described. This method involves the formation of new bond in solvent free aminolysis without the use of any additional reagents or catalyst and provided the product in good to excellent yield. The feasibility of activating reaction by the mean of temperature and pressure in microwave irradiation has been demonstrated. This eco-friendly procedure show notable advantages as: (a) operational simplicity; (b) fast reaction; (c) ecological advantages; (d) economic experience and (e) good yield.
References
- Horváth, I. T.; Anastas, P.T. Chem. Rev. 2007, 107, 2169-2173;
CrossRef - Liu, S.; Xiao, J. J. Mol. Catal. A: Chem. 2007, 270, 1-43;
CrossRef - Anastas, P.T. ; Zimmerman, J. B. Environ. Sci. Technol. 2003, 37, 94A-101A.
CrossRef - Bagley, M. C.; Chapaneri, K., Dale, J. W.; Xiong, X.; Bower, J. J. Org. Chem. 2005, 70, 1389-1399,
CrossRef - Griffin, R. J.; Fontana, G.; Golding, B. T.; Guiard, S.; Hardcastle, I. R.; Leahy, J. J.; Martin, N.; Richardson, C.; Rigoreau, L.; Stockley, M.; Smith, G. C. M. J. Med. Chem. 2005, 48, 569-585;
CrossRef - Takayama, K.; Iwata, M.; Hisamichi, H.; Okamoto, Y.; Aoki, M.; Niwa, A. Chem. Pharm. Bull. 2002, 50, 1050-1059;
CrossRef - Szczepankiewicz, B. G.; Heathcock, C. H. J. Org. Chem. 1994, 59, 3512-3513.
CrossRef - Meyer, C.; Piva, O.; Pete. J-P. Tetrahedron 2000, 56, 4479-4489.
CrossRef - Cossy, J.; Thellend, A. Synthesis 1989, 10, 753-755;
CrossRef - Miriyala, B.; Williamson, J. S. Tetrahedron Lett. 2003, 44, 7957-7959;
CrossRef - Štefane, B.; Polanc, S. Synlett. 2004, 4, 698-702;
CrossRef - Štefane, B.; Polanc, S. Tetrahedron 2007, 63, 10902-10913.
CrossRef - Witzeman, J. S.; Nottingham, W. D. J. Org. Chem. 1991, 56, 1713-1718.
CrossRef - Ley, S. V.; Woodward, P. R. Tetrahedron Lett. 1987, 28, 3019-3020.
CrossRef - Sung, K.; Wu, S. Y. Synth. Commun. 2001, 31, 3069-3074.
CrossRef - Johnson, D. H. J. Chem. Soc. 1958, 0, 1624-1628.
CrossRef - Bredereck, H.; Gompper, R.; Klemm, K. Chem Ber. 1959, 92, 1456-1460.
CrossRef - Clemens, R. J. Chem. Rev. 1986, 86, 241-318.
CrossRef - Gotor, V.; Liz, R.; Testera, A. M. Tetrahedron 2004, 60, 607-618.
CrossRef - Garcia, M. J.; Rebolledo, F.; Gotor, V. Tetrahedron Lett. 1993, 34, 6141-6142;
CrossRef - Ponde, D. E.; Deshpande, V. H.; Bullrile, V. J.; Sudalai, A.; Gajare, A. S. D. J. Org. Chem. 1998, 63, 1058-1063.
CrossRef - Neo, A. G.; Delgado, J.; Polo, C.; S. Marcaccini.; Marcos, C. F. Tetrahedron Lett. 2005, 46, 23-26.
CrossRef - Dudognon, Y.; Presset, M.; Rodriguez, J.; Bugaut, X.; Constantieux, T. Chem. Commun. 2016, 52, 3010-3013.
CrossRef - Presset, M.; Coquerel, Y.; Rodriguez, J. J. Org. Chem. 2009, 74, 415-418;
CrossRef - Leung-Toung, R.; Wentrup, C. J. Org. Chem, 1992, 57, 4850-485;
CrossRef - Cossy, J.; Leblanc, C. Tetrahedron Lett. 1991, 32, 3051-3052;
CrossRef - Wolff, L. Justus Liebigs Ann. Chem. 1912, 394, 86-108.
CrossRef

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


