Tandem Reactions Using Nitrile Imines: Synthesis of Some Novel Heterocyclic Compounds with Expected Biological Activity
Adil A. H. Gobouri1, Mosselhi A. N. M. Mohamed*1,2 and Mahmoud A. Amin1,3
1Department of Chemistry, Faculty of Science, Taif University, 21974-Taif/haweiah, Saudi Arabia. 2Department of Chemistry, Faculty of Science, Cairo University, 12613-Giza, Egypt. 3Department of Chemistry, Faculty of Science, Suez Canal University, Ismailia, Egypt. Corresponding Author Email: mosselhimohamed@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/320115
Article Received on :
Article Accepted on :
Article Published : 23 Feb 2016
New functionalized 7,9-dimethylpyrimido[4,5-d][1,2,4]triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione derivatives were synthesized via reaction of the hydrazonoyl halides with 7,8-dihydro-1,3-dimethyl-7-thioxopyrimido[4,5-d]pyrimidine-2,4,5(1H,3H,6H)trione. The biological activity of the products has been evaluated. The mechanism and the regioselectivity of the studied reactions have been discussed.
KEYWORDS:Tandem reaction; hydrazonoyl halides; nitrile imines; l,3-dipolar cycloaddition
Download this article as:| Copy the following to cite this article: Gobouri A. A. H, Mohamed M. A. N. M, Amin M. A. Tandem Reactions Using Nitrile Imines: Synthesis of Some Novel Heterocyclic Compounds with Expected Biological Activity. Orient J Chem 2016;32(1). |
| Copy the following to cite this URL: Gobouri A. A. H, Mohamed M. A. N. M, Amin M. A. Tandem Reactions Using Nitrile Imines: Synthesis of Some Novel Heterocyclic Compounds with Expected Biological Activity. Orient J Chem 2016;32(1). Available from: http://www.orientjchem.org/?p=14180 |
Introduction
Chemists are constantly working to discover new and improved reactions. One of the primary motivating goals of this research is the development of cleaner, more efficient transformations to shorten syntheses and save money on chemicals. The strategy of using reactions in tandem is also aimed at shortening syntheses. Tandem reactions are commonly referred to under the nebulous phrase “multistep one-pot
reactions”.1 On cost and amounts of reagents, solvents, and reduce the amount of waste that is generated. So, our aim is to use such idea of tandem reactions of organic compounds to synthesize new heterocycles with expected biological activity in short steps by using nitrile imines via 1,3-dipolar cycloaddition.
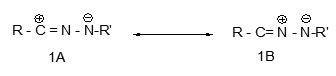
Basically a nitrile imine 1 is a flexible system of three atoms over which four pi-electrons are distributed. Although one can write seven possible resonance structures for such a system, the 1,3-dipolar sextet structure 1A with its complementary nucleophilic and electrophilic centers will be used throughout this article, although theoretical calculations have indicated that all the octet zwitterionic structure 1B is the most stable contributor to the resonance hybrid.2
As very authorative review of the chemistry of the precursors of nitrilimines as well as generation of nitrile imines was reported in 2010 by Shawali.2 Pyridopyrimidines have good biological importance.3 It acts as good pharmacophore.Recently some fused heterocyclic compounds containing nitrogen atom showa wide range of pharmacological activities.Pyrimido pyrimidines are annelated to uracils that have considerable interest in recent years.4,5 Derivatives of pyrimido pyrimidine display potent inhibitory properties regarding tyrosine kinase domain of epidermal growth factor receptor.6 Pyrimido[4,5-d]pyrimidine fused system represent attractive pharmacological applications such as antitumor,7 antiviral,8 antioxidant,9 antifungal10 and hepatoprotective activities.11 Pyrimido pyrimidines have a ring system that can be found marine derived natural products such as crambescidin 23 alkaloid. Keeping this importance in mind herein the reactions of 7-thioxopyrimdo[4,5-d]pyrimidin-2,4,5-trione with hydrazonoyl halides will be reported.
Experimental
General
All evaporations were carried out under reduced pressure at 60°C. TLC was carried out on aluminum sheet silica gel 60 (Fluka) and detected by UV light. All melting points were measured on an electrothermal melting point apparatus and are uncorrected. The 1H and 13C NMR spectra were recorded in deuterated dimethyl sulphoxide (DMSO-d6) at 300 MHz on a Varian Mercury VXR-300 NMR
Materials
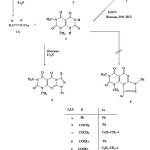
Synthesis of 7,8-dihydro-1,3-dimethyl-7-thioxopyrimido[4,5-d]pyrimidine-2,4,5(1H,3H,6H)trione (4)
A mixture of ethyl 6-amino-1,3-dimethyl-5-(ethoxycarbonyl)uracil (3)12 (11.35 g, 0.05 mole) and potassium thiocyanate (6.8 g, 0.07 mol) was stirred under reflux in dioxane (40 ml) containing 10% HCl (40 ml) for 20 hours. After cooling, the mixture was poured into 300 ml water, the solid precipitated was filtered off and crystallized from dioxane to give 4 as yellow powder.
Yield: 7.5 g, 62.5%; m.p. > 300 ◦C; IR ν 3150 (NH), 1705, 1680, 1630 (CO) cm−1; 1H NMR(DMSO-d6) δ 3.2 (s, 3H, CH3), 3.45 (s, 3H, CH3), 13.75 (s, 2H, NH); 13C NMR(DMSO-d6, 75 Mz) δ 29.0 (CH3), 30.5 (CH3), 85.0 (C4a), 150.5 (C2), 161.0 (C8a), 162.0 (C4), 163.5 (C5), 173.0 (C=S); MS, m/z (%) 240 (M+, 40); Anal. Calcd for C8H8N4O3S (240.2): C, 40.00; H, 3.36; N, 23.32l; S, 13.35. Found: C, 39.7; H, 3.2; N, 22.9; S, 13.0%.
Reactions of 4 with active chloromethylene compounds (10b,d,f)
To a solution of 4 (2.4 g, 0.01 mol) in chloroform was added triethylamine (1.4 ml, 0.01 mol) and the mixture was stirred for 10 min at room temperature. To the resulting clear solution was added active chloromethylene derivative (10) (0.01 mol) dropwise while stirring the reaction mixture. After complete addition, the reaction mixture was stirred for further 24 h at room temperature. The solid that precipitated was filtered off, washed with water, dried and finally crystallized from dioxane to give pure 11. The compounds 11b,d,fprepared are listed below together with their physical constants.
Dimethyl-2,4,5(1H,3H,6H)-trioxopyrimido[4,5-d]pyrimidin-7-yl] thio-2,4-dioxopentane (11b)
Yellow solid (2.0 g, 59%), m.p. 235 ◦C; IR (KBr) ν 3400 (NH), 1712, 1702, 1651, 1632 (CO) cm−1; 1H NMR (DMSO-d6) δ 2.0 (s, 6H, 2 COCH3), 3.20 (s, 3H, N-CH3), 3.4 (s, 3H, N-CH3), 4.7 (s, 1H, CH), 11.45 (s, 1H, NH); 13C NMR(DMSO-d6, 75 Mz) δ 20.2 (2CH3), 27.6 (CH3), 29.5 (CH3), 69.5 (CH), 95.8 (C4a), 144.5 (C8a), 145.78 (C2), 154.5 (C7-S), 161.5 (C4), 168.76 (C5), 172.13 (CH3CO); MS m/z (%) 338 (M+, 17); Anal. Calcd for C13H14N4O5S (338.34): C, 46.15; H, 4.17; N, 16.56; S, 9.48. Found C, 46.1; H, 3.9; N, 16.3; S, 9.5%.
Ethyl 1,3-dimethyl-2,4,5(1H,3H,6H)trioxopyrimido[4,5-d]-pyrimidin-7-yl] thio-3-oxobutanoate (11d)
Yellow solid (2.4 g, 65.0%), m.p. 244 ◦C ; IR (KBr) ν 3410 (NH), 1715, 1705, 1650, 1625 (CO) cm−1; 1H NMR (DMSO-d6) δ 1.31 (t, J = 7 Hz, 3H, CH3),2.6 (s, 3H, CH3), 3.26 (s, 3H, N-CH3), 3.45 (s, 3H, N-CH3), 4.30 (q, J = 7 Hz, 2H, CH2), 4.80 (s, 1H, CH), 12.8 (s, 1H, NH); 13C NMR(DMSO-d6, 75 Mz) δ 16.0 (CH3), 29.0 (CH3), 31.0 (CH3), 59.0 (CH2), 70.5 (CH), 95.0 (C4a), 148.0 (C8a), 153.8 (C2), 159.8 (C7-S), 161.0 (C4), 163.5 (C5), 179.0 (CH3CO), 189.5 (COOEt); MS m/z (%) 368 (M+, 35); Anal. Calcd for C14H16N4O6S (338.37): C, 45.65; H, 4.38; N, 15.21; S, 8.70. Found C, 45.5; H, 4.2; N, 15.0; S, 8.5%.
N-Phenyl 1,3-dimethyl-2,4,5(1H,3H,6H)-trioxopyrimido[4,5-d-pyrimidin-7-yl] thio-3-oxobutanamide (11f)
Yellow solid (3.0 g, 72.30%), m.p. 229 ◦C; (KBr) ν 3400, 3250 (2NH), 1725, 1672, 1652, 1630 (CO) cm−1; 1H NMR (DMSO-d6) δ 2.53 (s, 3H, CH3), 3.25 (s, 3H, N-CH3), 3.42 (s, 3H, N-CH3), 4.6 (s, 1H, CH), 7.27-7.50 (m, 5H, ArH), 9.0 (s, 1H, NH), 10.1 (s, 1H, NH); 13C NMR(DMSO-d6, 75 Mz) δ 20.0 (CH3), 28.0 (CH3), 29.7 (CH3), 68.5 (CH), 94.5 (C4a), (120.0, 126.5, 128.5, 130.0 C-Ar.), 148.0 (C8a), 152.0 (C2), 161.0 (C7-S), 161.5 (C4), 164.0 (C5), 165.8 (NHCO), 179.5 (COCH3). MS m/z (%) 415 (M+, 40); Anal.Calcd for C18H17N5O5S (415.42): C, 52.04; H, 4.12; N, 16.86; S, 7.72. Found C, 52.0; H, 3.9; N, 16.5; S, 7.5%.
Synthesis of the thiohydrazonates (7)
To a solution of each of 11b, 11d and 11f (10 mmol) in ethanol (40 ml) was added sodium acetate trihydrate (1.38 g, 10 mmol) and the mixture was cooled to 0–5 ◦C in an ice bath. To the resulting cold solution was added portionwise a cold solution of benzenediazonium chloride, prepared by diazotizing aniline (10 mmol) dissolved in hydrochloric acid (6 M, 6 ml) with a solution of sodium nitrite (0.7 g, 10 mmol) in water (10 ml). After complete addition of the diazonium salt, the reaction mixture was stirred for further 12 h at room temperature. The solid precipitated was filtered off, washed with water, dried and crystallized from dimethyformamide/EtOH (1:1 v:v) to give the respective pure 7b, 7d and 7f.
Dimethyl-2,4,5(1H,3H,6H)-trioxopyrimido[4,5-d]pyrimidin-7-yl]-N-phenyl-2-oxopropanethiohydrazonate (7b).
Yellow solid (3.0 g, 75.0%), m.p. 298 ◦C; (KBr) ν 3350, 3235 (2NH), 1710, 1660, 1630, 1590 (CO) cm−1; 1H NMR (DMSO-d6) δ 2.50 (s, 3H, CH3), 3.26 (s, 3H, N-CH3), 3.45 (s, 3H, N-CH3), 7.27-7.40 (m, 5H, ArH), 9.45 (s, 1H, NH), 10.2 (s, 1H, NH); MS m/z (%) 400 (M+, 20); Anal. Calcd for C17H16N6O4S (400.41): C, 50.99; H, 4.03; N, 20.99; S, 8.01. Found C, 50.6; H, 3.9; N, 20.7; S, 8.0%.
Dimethyl-2,4,5(1H,3H,6H)-trioxopyrimido[4,5-d]pyrimidin-7-yl]-N-phenyl-2-ethoxy-3-oxo-ethanethiohydrazonate (7d).
Yellow solid (3.2 g, 74.4%), m.p. > 300 ◦C; (KBr) ν 3390, 3255 (2NH), 1705, 1680, 1640, 1585 (CO) cm−1; 1H NMR (DMSO-d6) δ 1.33 (t, J = 7 Hz, 3H), 3.25 (s, 3H, N-CH3), 3.45 (s, 3H, N-CH3), 4.49 (q, J = 7 Hz, 2H), 7.2-7.70 (m, 5H, ArH), 10.2 (s, 1H, NH), 11.0 (s, 1H, NH); MS m/z (%) 430 (M+, 33); Anal. Calcd for C18H18N6O5S (430.44): C, 50.23; H, 4.21; N, 19.52; S, 7.45. Found C, 50.1; H, 3.89; N, 19.3; S, 7.2%.
Dimethyl-2,4,5(1H,3H,6H)-trioxopyrimido[4,5-d]pyrimidin-7-yl]-N-phenyl-2-(phenylamino)-2-oxo-ethanethiohydrazonate (7f).
Yellow solid (3.5 g, 73.3%), m.p. > 300 ◦C; (KBr) ν 3350, 3245 (2NH), 1707, 1670, 1645, 1595 (CO) cm−1; 1H NMR (DMSO-d6) δ 3.26 (s, 3H, N-CH3), 3.45 (s, 3H, N-CH3), 7.3-7.5 (m, 10H, ArH), 9.0 (s, 1H, NH), 9.35 (s, 1H, NH), 9.9 (s, 1H, NH); MS m/z (%) 477 (M+, 48); Anal. Calcd for C22H19N7O4S (477.5): C, 55.34; H, 4.01; N, 20.53; S, 6.72. Found C, 55.1; H, 3.7; N, 20.2; S, 6.5%.
Synthesis of 7,9-dimethylpyrimido[4,5-d][1,2,4]triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5a-g).
Method A
To a mixture of 7,8-dihydro-1,3-dimethyl-7-thioxopyrimido[4,5-d]pyrimidine-2,4,5(1H,3H,6H)trione (4) (2.4 g, 0.01 mol) and appropriate hydrazonoyl halide 1 (0.01 mol) in dioxane (40 ml), trimethylamine (1.4 ml, 0.01 mol) was added. The reaction mixture was refluxed under stirring till hydrogen sulfide ceased to evolve (6-10 h). The solvent was evaporated and the residue was treated with ice/HCl mixture. The solid product was collected, washed with water and crystallized from proper solvent to give the respective 5 in 50–60% yield.
Method B (for 5b, 5d and 5f)
To a stirred ethanolic sodium ethoxide solution, prepared from sodium metal (0.23 g, 10 mg atom) and absolute ethanol (20 ml), was added each of the compound 7b, 7d and 7f (10 mmol) and the reaction mixture was stirred at room temperature for 15 h, during which the starting reactants 7 dissolved and the crude product preciptated. The latter was filtered, washed with water, dried and finally crystallized from the proper solvent to give a product identified as 5b, 5d and 5f, respectively. The latter products proved to be identical in all respects (mp, mixed mp, IR) with that obtained from 4 and the respective hydrazonoyl halides 1. Their yields were 60–70%.
Dimethyl-1,3-diphenylpyrimido[4,5-d][1,2,4]triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5a)
Pale yellow solid (2.3 g, 57.5%), m.p. > 300 ◦C; (KBr) ν 1695, 1630 (CO) cm−1; 1H NMR (DMSO-d6) δ 3.28 (s, 3H, N-CH3), 3.40 (s, 3H, N-CH3), 7.27-8.0 (m, 10H, ArH); 13C NMR (DMSO-d6, 75 MHz) δ 27.2 (CH3), 29.3.0 (CH3), 94.2 (C5a), 127.2, 127.3, 128.38, 130.49, 131.85, 132.42, 136.73 (C-Ar), 144.0 (N1-C), 148.0 (C9a), 150.0 (C8), 160.0 (C3), 161.5 (C5), 163.0 (C6); MS m/z (%) 400 (M+, 19); Anal. Calcd for C21H16N6O3 (400.39): C, 62.99; H, 4.03; N, 20.99. Found C, 62.75; H, 3.9; N, 20.7%.
Acetyl-7,9-dimethyl-1-phenylpyrimido[4,5-d][1,2,4]triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5b)
Pale yellow solid (2.2 g, 60.1%), m.p. > 300 ◦C; (KBr) ν 1710, 1690, 1640 (CO) cm−1; 1H NMR (DMSO-d6) δ 2.5 (s, 3H, CH3), 3.1 (s, 3H, N-CH3), 3.4 (s, 3H, N-CH3), 7.25-7.85 (m, 5H, ArH); 13C NMR (DMSO-d6, 75 MHz) δ 21.0 (CH3), 33.49 (CH3), 34.28 (CH3), 90.9 (C5a), 117.0, 119.0, 123.0, 125.5, 127.5, 132.0, (C-Ar), 138.5 (N1-C), 150.0 (C9a), 155.2 (C8), 161.5 (C3), 162.5 (C5), 163.6 (C6), 172.0 (COCH3); MS m/z (%) 366 (M+, 35); Anal. Calcd for C17H14N6O4 (366.33): C, 55.74; H, 3.85; N, 22.94. Found C, 55.5; H, 3.6; N, 22.65%.
Acetyl-7,9-dimethyl-1-(4-methylphenyl)pyrimido[4,5-d][1,2,4]-triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5c)
Pale yellow solid (2.3 g, 60.5%), m.p. > 300 ◦C; (KBr) ν 1710, 1685, 1635 (CO) cm−1; 1H NMR (DMSO-d6) δ 2.5 (s, 3H, CH3), 2.6 (s, 3H, CH3), 3.27 (s, 3H, N-CH3), 3.40 (s, 3H, N-CH3), 7.4-7.6 (d, J = 8 Hz, 2H, ArH), 7.7-8.09 (d, J = 8 Hz, 2H, ArH); MS m/z (%) 380 (M+, 30); Anal. Calcd for C18H16N6O4 (380.36): C, 56.84; H, 4.24; N, 22.10. Found C, 56.8; H, 4.0; N, 22.0%.
Dimethyl-3-ethoxycarbonyl-1-phenylpyrimido[4,5-d][1,2,4]-triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5d)
Pale yellow solid (2.2 g, 55.5%), m.p. 299 ◦C; (KBr) ν 1730, 1695, 1650 (CO) cm−1; 1H NMR (DMSO-d6) δ 1.3 (t, J = 7 Hz, 3H), 3.32 (s, 3H, N-CH3), 3.55 (s, 3H, N-CH3), 4.35 (q, J = 7 Hz, 2H), 7.25-7.80 (m, 5H, ArH); 13C NMR (DMSO-d6, 75 MHz) δ 28.1 (CH3), 29.88 (CH3), 31.5 (CH3), 59 (CH2), 92.9 (C5a), 116.5, 119.0, 123.6, 125.8, 126.3, 127.6, 131.0, 132.5, (C-Ar), 144.0 (N1-C), 151.0 (C9a), 154.5 (C8), 161.0 (C3), 162.2 (C5), 163.3 (C6), 174.0 (COOEt); MS m/z (%) 396 (M+, 15); Anal. Calcd for C18H16N6O5 (396.36): C, 54.54; H, 4.07; N, 21.2. Found C, 54.3; H, 3.7; N, 21.0%.
7,9-Dimethyl-3-ethoxycarbonyl-1-(4-methylphenyl)pyrimido[4,5-d][1,2,4]-triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5e)
Pale yellow solid (2.1 g, 51.0%), m.p. 267 ◦C; (KBr) ν 1735, 1700, 1645 (CO) cm−1; 1H NMR (DMSO-d6) δ 1.3 (t, J = 7 Hz, 3H), 2.6 (s, 3H, CH3), 3.27 (s, 3H, N-CH3), 3.45 (s, 3H, N-CH3), 4.4 (q, J = 7 Hz, 2H), 7.20-7.45 (d, J = 8 Hz, 2H, ArH), 7.6-7.8 (d, J = 8 Hz, 2H, ArH); MS m/z (%) 410 (M+, 25); Anal. Calcd for C18H16N6O5 (410.38): C, 55.61; H, 4.42; N, 20.48. Found C, 55.5; H, 4.1; N, 20.3%.
Dimethyl-3- phenylaminocarbonyl-1-phenylpyrimido[4,5-d][1,2,4]-triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5f)
Yellow solid (2.2 g, 50.0%), m.p. > 300 ◦C; (KBr) ν 3255 (NH), 1705, 1675, 1630 (CO) cm−1; 1H NMR (DMSO-d6)δ 3.25 (s, 3H, N-CH3), 3.40 (s, 3H, N-CH3), 7.25-7.80 (m, 10H, ArH), 10.6 (s, 1H, NH); 13C NMR (DMSO-d6, 75 MHz) δ 33.0 (CH3), 39.0 (CH3), 91.0 (C5a), 127.5, 130.0, 132.5, 138.0 (C-Ar), 144.0 (N1-C), 150.5 (C9a), 154.0 (C8), 161.5 (C3), 162.5 (C5), 163.9 (C6); MS m/z (%) 443 (M+, 27); Anal. Calcd for C22H17N7O4 (443.41): C, 59.59; H, 3.86; N, 22.11. Found C, 59.3; H, 3.5; N, 21.8%.
Dimethyl-3- phenylaminocarbonyl-1-(4-methylphenyl)-pyrimido[4,5-d][1,2,4]-triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5g)
Yellow solid (2.5 g, 54.7%), m.p. > 300 ◦C; (KBr) ν 3250 (NH), 1705, 1665, 1615 (CO) cm−1; 1H NMR (DMSO-d6) δ 3.26 (s, 3H, N-CH3), 3.45 (s, 3H, N-CH3), 7.25-8.0 (m, 9H, ArH), 10.31 (s, 1H, NH); MS m/z (%) 457 (M+, 30); Anal. Calcd for C23H19N7O4 (457.44): C, 60.39; H, 4.19; N, 21.43. Found C, 60.0; H, 4.0; N, 21.1%.
Antimicrobial assay
Cultures of four fungal species namely Aspergillus fumigatus AF, Penicillium italicum PI, Syncephalastrum racemosum SR and Candida albicans. CA as well as four bacterial species namely Staphylococcus aureus SA, Pseudomonas aeruginosa PA, Bacillus subtilis BS and Escherichia coli EC were used to investigate the antimicrobial activity of the compounds 5a-g. The antimicrobial activity was assayed biologically using the diffusion plate technique. The latter technique was carried out by pouring a spore suspension of the fungal species (one ml of sterile water contains approximately 108 conidia) or spreading bacterial suspension over a solidified malt agar medium. The layer is allowed to set for 30 min. A solution of the test compound 5a-g (1.0μg/ml) in dimethylformamide was placed onto sterile 5mm filter paper discs and allowed to dry, then the discs were placed on the centre of the malt agar plate and incubated at optimum incubation temperature 28 ± 2◦C. Test organism growth may be affected by the inhibitory action of the test compound, so a clear zone around the disc appears as an indication of the inhibition of test organism growth. The size of the clearing zone is proportional to the inhibitory action of the compound. The fungicide Terbinafin and the bactericide Chloramphenicol were used as standards under the same conditions. Measurements were considered after 72 h for fungi and 24 h for bacteria. The results are summarized in table 1.
Results and Discussion
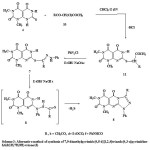
The starting 7,8-dihydro-1,3-dimethyl-7-thioxopyrimido[4,5-d]pyrimidine-2,4,5(1H,3H,6H)trione (4) which has not been reported hitherto was prepared by heating of 6-amino-1,3-dimethyluracil (2) with ethyl chloroformate in pyridine to get ethyl 6-amino-1,3-dimethyl-5-(ethoxycarbonyl)uracil (3),12 followed by reaction with potassium thiocyanate in 1,4-dioxane containing 10% HCl.13 The structure of compound 4 was confirmed by spectral and elemental analysis data (see Experimental). The hydrazonoyl halides 1a–g14-16 were prepared by literature methods. Reaction of 4 with each of the hydrazonoyl halides 1a–g was carried out in 1,4-dioxane in the presence of triethylamine while heating the reaction mixture under reflux till in all cases, hydrogen sulfide evolved during the course of the reaction and so stirring of the reaction mixture was continued till hydrogen sulfide ceased to evolve (6-10 h). Work up of the reaction mixture afforded, in each case, one isolable product as evidenced by tlc analysis of the crude product. On the basis of elemental analyses and IR, 1H and 13C NMR spectra which showed all the expected signals (see Experimental), the isolated products were assigned the structure of 7,9-dimethylpyrimido[4,5-d][1,2,4]triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione (5) rather than the isomeric structure of 1,3-dimethylpyrimido[5,4-e][1,2,4]triazolo[4,3-a]pyrimidine-2,4,5(1H,3H,7H)-trione (6) (scheme 1). For example, the δ value (163.0) for the carbonyl carbon signal in the 13C nmr spectrum of 5a, taken as an example of the series prepared is similar to that of I (δ161–164) and different from its isomeric structure II (δ170–175)14 (chart 1). This finding ruled out the acylimino type structure 5 for the isolated products.
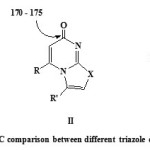 |
Chart 1: 13C comparison between different triazole derivatives |
 |
Scheme 1 Click here to View scheme |
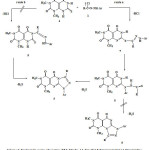
The formation of compounds 5 from the thione 2 and hydrazonoyl halides 1 could be accounted for by one of the two pathways indicated in scheme 2. As depicted in this scheme, it is suggested that the studied reactions started with the hydrazonoylation of 2 to give the respective thiohydrazonate esters 7. This is followed by Smiles type rearrangement17,18 of 7 to form the respective thiohydrazides 8, which in turn underwent cyclization to give 5 as end products (route a, Scheme 2).
It seems that both intermediates 7 and 8 are consumed, under the employed reaction conditions as soon as they are formed since all attempts to isolate them failed. Alternatively, reaction of the thione 2 with hydrazonoyl halides 1 starts with the formation of the amidrazone intermediates 9 which cyclize to give 5 (route b, Scheme 1). This alternative pathway has been ruled out, however, on the basis that alkylation and acylation of 2-thiouracil derivatives have been known to give S-alkyl and S-acyl derivatives, respectively.19-22 Furthermore the suggested route a and the involvement of 7 and 8 as intermediates in the formation of 4 by this route were evidenced by alternate synthesis of 5b, 5d and 5f (Scheme 3).
 |
Scheme 2 Click here to View scheme |
Thus, treatment of 2 with each of 3-chloro-2,4-pentanedione (10b), ethyl α-chloroacetoacetate (10d) and α-chloroacetoacetanilide (10f) in chloroform in the presence of triethylamine afforded the respective substituted products 11b, 11d and 11f. Coupling of each of these compounds with benzenediazonium chloride in ethanol in the presence of sodium acetate yielded the thiohydrazonates 7b, 7d and 7f, respectively (Scheme 2) via Japp-Klingemann cleavage of the acetyl group.22 Treatment of the products 7 with sodium ethoxide in ethanol afforded the respective compounds 5b, 5d and 5f identical in all respects with those obtained from reactions of 2 with each of 1b, 1d and 1f, respectively. This finding indicates that 7 and 8 are intermediates in the studied reactions of 1 with 3 and they are consumed as soon as they are formed under the employed reaction conditions. Finally, the suggestion that the site of cyclization of the thiohydrazide intermediates 8 involves N-3 rather than N-1 to give 5 is consistent with literature reports. For example, it has been reported that cyclization of 2-substituted-uracil derivatives having no substituent on N-3 proceeds regioselectively to give the respective 1,2,4-triazolo[4,3-a]pyrimidin-5(1H)- ones.23-25 In conclusion, the studied reactions of hydrazonoyl halides 1 with the thione 2 are both site and regioselective and lead to the title ring system.
 |
scheme 3 Click here to View scheme |
Antimicrobial activity
The compounds 5a-g were tested for their antimicrobial activities against four fungal species namely Aspergillus fumigatus AF, Penicillium italicum PI, Syncephalastrum racemosum SR and Candida albicans CA as well as four bacteria species namely Staphylococcus aureus SA, Pseudomonas aeruginosa PA, Bacillus subtilis BS and Escherichia coli EC. The organisms were tested against the activity of solutions of
Table 1: Antimicrobial activity of the products 5a-g.
|
Micro-Organism/IZD(cm)f
|
||||||||
|
Compd. No. |
AF |
PI |
SR |
CA |
SA |
PA |
BS |
EC |
|
5a |
++ |
+ |
0 |
+ |
++ |
0 |
0 |
0 |
|
5b |
+ |
0 |
+ |
0 |
++ |
0 |
+ |
0 |
|
5c |
+ |
0 |
0 |
0 |
+ |
0 |
+ |
0 |
|
5d |
+ |
0 |
0 |
0 |
++ |
+ |
0 |
0 |
|
5e |
+ |
+ |
0 |
0 |
+ |
+ |
0 |
+ |
|
5f |
+ |
0 |
0 |
+ |
+ |
+ |
0 |
0 |
|
5g |
++ |
0 |
0 |
+ |
+ |
0 |
+ |
0 |
|
CAa |
++ |
++ |
++ |
++ |
||||
|
TEb |
++ |
++ |
++ |
++ |
||||
f50 ml of solution in DMF whose concentration 1.0m g/ml was tested.
a Chloramphenicol as standard antibacterial agent, b Terbinfin as standard antifungal agent, ++ inhibition value 0.6-1.0 cm; +, inhibition value 0.1-0.5 cm beyond control; 0, no inhibition detected.
concentration of 1.0μg/ml of each compound and using inhibition zone diameter in cm (IZD) as criterion for the antimicrobial activity. Terbinafin as an antifungal agent and Chloramphenicol as an antibacterial agent were used as references to evaluate the potency of the tested compounds under the same conditions. The results are depicted in table 1. The results reveal that compounds 5a, 5b, 5d and 5g exhibited the highest degree of inhibition against the tested organisms AF and SA, respectively, their activity is similar to that of the standard antifungal and antibacterial agents used. All other compounds either exhibit no activity or being less active against the tested species.
Conclusion
The present project has outlined the importance of tandem in situ generation and 1,5-electrocyclization of N-hetaryl nitrilimines as a convenient methodology for synthesis of numerous 1,2,4-triazole derivatives, namely functionalized 7,9-dimethylpyrimido[4,5-d][1,2,4]triazolo[4,3-a]pyrimidine-5,6,8(1H,7H,9H)-trione derivatives. Some compounds prepared showed moderate, whereas other compounds showed weak antimicrobial activity.
Acknowledgement
The author thanks Taif University, Taif, Saudi Arabia for the financial support under project number 3674-435-1
References
- Cava, M.P.;Pollack, N.M.;Dieterle, G.A. J. Am. Chem. Soc., 1973, 95, 2558-2560.
CrossRef - Ho, T., Tandem Organic Reactions; Wiley-Interscience; New York 1992.
- Shawali, A. S., ARKIVOC 2010, (i), 33-97.
- Pedgaonkar, Y. Y.; Leie, A. C.; Desai, N. H. P.; Degani, M. S.; Int. J. Pharm. Bio. Sci. 2014, 5, 422-439 .
- Clark, A., Pharm Res. 1996, 13, 1133-1141.
CrossRef - Melik-Ogandzhanyan, R. G.; Khachatryan, V. E.; Gapoyan A. S., Russ Chem Rev. 1985, 4, 262-276.
CrossRef - Rewcastle, G. W.; Bridge, A. J.; Fry, D. W.; Rubin, J. R.; Denny, W. A., J. Med. Chem. 1997, 40, 1820-1826.
CrossRef - Sanghvi, Y. S.; Larson, S. B.; Matsumoto, S. S.; Nord, L. D.; Smee, D. F.; Willis, R. C.; Avery, T. H.; Robins, R. K.; Revankar, G. R., J. Med. Chem. 1989, 32, 629-637.
CrossRef - Tenser, R. B.; Gaydos, A. K.; Hay, A., Antimicrob Agents Chemother., 2001, 45, 3657-3659.
CrossRef - De la Cruz, J.P.; Carrasco, T; Ortega, G; Sanchez, De la; Cuesta, F., Lipids 1992, 27, 192-194.
CrossRef - Sharma, P.; Rane, N.; Gurram, V. K.; Bioorg. Med. Chem. Lett. 2004, 14, 4185- 4190.
CrossRef - Ram, V. J.; Goel, A.; Sarkhel, S.; Maulik, P. R., Bioorg. Med. Chem. 2002, 10, 1275-1280.
CrossRef - Yoneda, F.; Higuchi, M., Bull. Chem. Soc. Japan 1973, 46, 3849-3853.
CrossRef - EL-Gazzar, A.-R. B. A.; Hussein, H. A. R.; Hafez, H. N., Acta Pharm. 2007, 57, 395-411.
CrossRef - Shawali, A. S.; Elghandour, A. H.; Sayed. A. R., Synthetic Commun. 2001, 31, 731-740.
CrossRef - Mosselhi, M. A. N.; Tawfik, N. M.; Shawali, A. S., Monatsch. Chem. 2003, 134, 565-571.
CrossRef - Shawali, A. S., Mosselhi, M. A. N., Tawfik, N. M., J. Org. Chem. 2001, 66, 4055-4057.
CrossRef - Ishii, K., Hatanaka, M., Ueda, I., Chem. Pharm. Bull. 1991, 39, 3331-3334.
CrossRef - Bunnett., J. F., Quart. Rev. 1958, 12, 1-16.
- Hurst, D. T., Beaumont, C., Jones, D. T. E., Kingsley, D. A., Partridge, J. D., Rutherford, T. J., Aust. J. Chem. 1988, 41, 1209-1219.
CrossRef - Sodereviciute, V., Vainilavicius, P., Chemija 1993, 2, 70.
- Ghoneim, K. M., El-Telbany, F. A., El-Enany, M., Youssef, K., Egypt. J. Pharm. Sci. 1987, 28, 127-136.
- Geies, A. A., Kamal-Eldeen, A. M., Abdelhafez, A. A., Gaber, A. M., Phosphorus, Sulfur & Silicon 1991, 56, 87-93.
CrossRef - Phillip, R. R., In Organic Reactions, R. Adams (Ed.), JohnWiley Sons, NewYork 1959, 10, chap.2, p. 143,
- Abdel-Aziz, S. A., Allimony, H. A., ElShaar, H. M., Usama, F. A., Abdel- Rahman, R. M., Phosphorus, Sulfur & Silicon 1996, 113, 67-77.
- Abdelfattah, A. M., Negm, A. M., Gaafar, A. E., Phosphorus, Sulfur & Silicon 1992, 72, 145-156.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


