Reductive ring opening of 3,5-bis(2-arylethenyl)isoxazoles with molybdenum hexacarbonyl: A novel route to symmetrical and unsymmetrical curcumin derivatives
Viwat Hahnvajanawong*, Thanaphat Thaima, Ruchanok Tearavarich and Parinya Theramongkol
Department of Chemistryand Center for Innovation in Chemistry, Faculty of Science, KhonKaen University, KhonKaen 40002, Thailand
DOI : http://dx.doi.org/10.13005/ojc/320113
Article Received on :
Article Accepted on :
Article Published : 27 Jan 2016
Curcumin derivatives were successfully synthesized from 3,5-dimethylisoxazole by lateral metalation and condensation with various aromatic aldehydes sequentially at C5- and C3-methyl groups. After dehydration, further transformation of isoxazole ring to b-diketone moiety was accomplished by reductive ring opening using molybdenum hexacarbonyl [Mo(CO)6] and subsequent simple acidic hydrolysis.
KEYWORDS:curcuminderivatives; reductive ring opening; molybdenum hexacarbonyl; lateral metalation
Download this article as:| Copy the following to cite this article: Hahnvajanawong V, Thaima T, Tearavarich R, Theramongkol P. Reductive ring opening of 3,5-bis(2-arylethenyl)isoxazoles with molybdenum hexacarbonyl: A novel route to symmetrical and unsymmetrical curcumin derivatives. Orient J Chem 2016;32(1). |
| Copy the following to cite this URL: Hahnvajanawong V, Thaima T, Tearavarich R, Theramongkol P. Reductive ring opening of 3,5-bis(2-arylethenyl)isoxazoles with molybdenum hexacarbonyl: A novel route to symmetrical and unsymmetrical curcumin derivatives. Orient J Chem 2016;32(1). Available from: http://www.orientjchem.org/?p=13750 |
Introduction
Curcumin, 1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadien-3,5-dione, is a bioactive compound isolated from Curcuma longarhizomes. Curcumin has been reported to possessantioxidant,anti-inflammatory, antitmicrobial and anticarcinogenic activities.1Variouscurcuminderivatives have been synthesized for testing their biological activities. Synthetic symmetrical curcuminderivativesreported in literature2-5 were obtained from Pabons procedure.6 Unsymmetrical curcuminderivatives, on the other hand, were synthesized by solid-phase synthetic strategy.7 Both syntheses proceeded through acetylacetone-boric anhydride complex which methyl terminals of this complex were allowed to undergo aldol condensation.
Isoxazoles can be employed as masked 1,3-dicarbonyl compounds because reductive cleavage of the N-O bond in isoxazoles produce b-aminoenones which are readily transform to their corresponding b-hydroxyenones. We have envisioned for using this N-O bond cleavage in our strategy for the synthesis of symmetrical as well as a wide range of unsymmetrical curcuminderivatives1, i.e., using isoxazole rings of 3,5-bis(2-arylethenyl)isoxazoles2 as masked 1,3-dicarbonyl moieties of curcuminderivatives1(Scheme 1). The synthetic direction to obtain 2 involves transformation of methyl groups of 3,5-dimethylisoxazole (5) into 2-aryl-2-hydroxyethyl groups sequentially at C5 and C3to get5-(2-aryl-2-hydroxyethyl)-3-methylisoxazoles 4and 3,5-bis(2-aryl-2-hydroxyethyl)isoxazoles3 respectively. Subsequent dehydration of 3,5-bis(2-aryl-2-hydroxyethyl)isoxazoles3 provides the route to 3,5-bisstyrylisoxazoles 2.
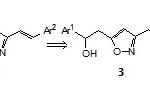 |
Scheme 1 Click here to View scheme |
Experimental Section
General
Melting points were determined by using a Sanyo Gallenkamp melting point apparatus and are uncorrected. IR spectra were taken with a Perkin Elmer Spectrum One FT-IR Spectrometer. 1H and 13C NMR spectra were recorded using a VARIAN MERCURY plus (400 MHz FT NMR).
n-BuLiand s-BuLi were prepared and their concentration were determined following the standard procedure.8,9
Preparation of 3,5-dimethylisoxazole (5)10
A solution of hydroxylamine hydrochloride (8.34 g, 0.12 mole) in water (10 ml) was added a solution of 2,4-pentanedione (6) (10.2 g, 0.1 mole) in ethanol (10 ml). The mixture was heated under reflux temperature for 3 hours, then allowed to cool to room temperature and poured into cold water (60 ml). The resulting mixture was extracted with ether (3×40 ml). The combined organic layer was dried (Na2SO4)and evaporated to drynessunder reduced pressure. The brown oily residue was purified by distilling under reduce pressure to give 3,5-dimethylisoxazole (5) (8.10g, 82%) as a colourless liquid (bp85-86°C at 150 mmHg);IR (thin film) nmax: 3132, 2934, 1611, 1455, 1414, 1258, 1011, 886, 795 cm-1; 1H NMR (CDCl3, 400 MHz) d: 5.82 (1H, s, H-4), 2.36 (3H, s, C=CCH3), 2.23 (3H, s, N=CCH3); 13C NMR (CDCl3) d: 11.3, 12.1, 102.3, 159.9, 169.1.
General procedure for preparation of 5-(2-aryl-2-hydroxyethyl)-3-methylisoxazoles 4a-c
To a stirred solution of 3,5-dimethylisoxazole (5) (2.91 g, 30.0 mmol) in dry THF (60 ml) was added n-BuLi (1.0 M in hexane; 30 ml, 30.0 mmol) dropwise under N2 at -78 °C and the mixture was stirred for an additional1 hour.A solution of aromatic aldehyde (30.0 mmol) in THF (6 ml) was then added. The resulting mixture was allowed to warm up to room temperature and treated with water. The phases were separated and the aqueous layer was extracted with ethyl acetate (3´120 ml).The combined organic layer was dried (Na2SO4) and concentrated under reduce pressure. Purification of the residue using column chromatography on silica gel with a gradient of 10-50% ethyl acetate in hexane as eluent gave5-(2-aryl-2-hydroxyethyl)-3-methylisoxazoles4a-c (70-84%) as yellow oils or as white crystalline solids.
5-(2-phenyl-2-hydroxyethyl)-3-methylisoxazole (4a)
Yield: 4.69 g (77%); yellow oil; IR (thin film) nmax:3370, 3031, 2931, 1604, 1493, 1450, 1415,1051, 742, 700 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.33-7.20 (5H, m,ArH), 5.80 (1H, s, =CH), 4.96 (1H, dd, J = 8.2 and 5.1 Hz, OCH ), 3.10 (1H, dd, J = 15.2 and 8.2 Hz, HCH), 3.01 (1H, dd, J = 15.2 and 5.0 Hz, HCH), 2.12 (3H, s, CH3);13C NMR (CDCl3) δ: 11.3, 36.6, 72.1, 103.4, 125.7, 128.0, 128.6, 143.0, 159.8, 169.6.
5-[2-(4-methoxyphenyl)-2-hydroxyethyl]-3-methylisoxazole (4b)
Yield: 4.90 g (70%); yellow oil; IR (thin film) nmax: 3376, 2934, 2838, 1608, 1513, 1416, 1246, 1177, 1032, 833 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.21 (2H, d, J = 8.7 Hz, ArH), 6.82 (2H, d, J = 8.6 Hz, ArH), 5.80 (1H, s, =CH), 4.92 (1H, dd, J = 8.0 and 5.4 Hz, OCH), 3.74 (3H, s, OCH3), 3.10 (1H, dd, J = 15.2 and 8.1 Hz, HCH), 2.99 (1H, dd, J = 15.2 and 5.3 Hz, HCH), 2.15 (3H, s, CH3); 13C NMR (CDCl3) δ: 11.2, 36.5, 55.2, 71.4, 103.2, 113.8, 127.0, 135.4, 159.1, 159.7, 169.9.
5-[2-(3,4-dimethoxyphenyl)-2-hydroxyethyl]-3-methylisoxazole(4c)
Yield: 6.63 g (84%); white crystals; mp 63-64 °C; IR (thin film) nmax: 3382, 2936, 1603, 1513, 1416, 1260, 1233, 1137, 1023, 809, 763 cm-1; 1H NMR (CDCl3, 400 MHz) d: 6.90-6.78 (3H, m, ArH), 5.83 (1H, s, =CH), 4.98 (1H, m, OCH), 3.83 (6H, s, 2´OCH3), 3.14 (1H, dd, J = 15.1 and 8.2 Hz, HCH), 3.04 (1H, dd, J = 15.1 and 5.1 Hz, HCH), 2.20 (3H, s, CH3); 13C NMR (CDCl3) δ: 11.3, 36.7, 55.9, 71.7, 103.5, 109.1, 111.2, 118.1, 136.1, 148.6 149.1, 160.0, 170.0.
General procedure for preparation of 3,5-bis(2-aryl-2-hydroxyethyl)isoxazoles3a-f
To a solution of 5-(2-aryl-2-hydroxyethyl)-3-methylisoxazole(4.0 mmol) in THF (20 ml) was added s-BuLi (1.0 M in hexane, 10.0 ml, 10.0 mmol) dropwise under N2 at -78 °C.After being stirred at -78 °C for 1 hour, a solution of aromatic aldehyde (5.0 mmol) in THF (2ml) was then added. The reaction mixture was allowed to warm up to room temperature and treated with water. The phases were separated and the aqueous layer was extracted with ethyl acetate (3 x 50 ml). The combined organic layer was dried (Na2SO4) and concentrated under reduced pressure. Purification of the residue using column chromatography on silica gel with agradient of 30-70% ethyl acetate in hexane as an eluent gave3,5-bis(2-aryl-2-hydroxyethyl)isoxazoles 4a-f (46-65%) as crystalline solids or as oils.
3,5-bis(2-phenyl-2-hydroxyethyl)isoxazole (3a)
Yield: 0.59 g (48%); white crystals; mp 97-98°C; IR (thin film) nmax: 3377, 3031, 1601, 1428, 1052, 754, 700 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.38-7.16 (10H, m, ArH), 5.89 and 5.88 (1H, 2s, =CH), 5.03-4.84 (2H, m, 2″-and2′-OCH), 3.09 (1H, dd,J = 15.2 and 8.7 Hz, 1″-HCH), 3.02 (1H, dd, J = 15.2 and 4.6 Hz, 1″-HCH), 2.98-2.89 (2H, m, 1′-CH2); 13C NMR (CDCl3) δ: 36.0, 36.7, 71.9, 72.4, 103.3, 125.7, 125.8, 127.8, 128.0, 128.5, 128.6 , 142.9, 143.2, 161.4, 169.9.
3,5-bis(2-(4-methoxyphenyl)-2-hydroxyethyl)isoxazole(3b)
Yield: 0.72 g (49%); white crystals;mp 97-99 °C; IR (thin film) nmax: 3377, 2957, 1608, 1514, 1253, 1175, 1033, 825 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.27 (4H, d, J = 8.4 Hz, ArH), 6.88(4H, d, J = 8.4 Hz, ArH) 5.88 and 5.87 (1H, 2s, =CH), 5.03-4.91(2H, m, 2″-and2′-OCH), 3.80 (6H, s, 2´OCH3), 3.16 (1H, dd, J = 15.2 and 8.4 Hz, 1″-HCH),3.10-2.92 (3H, m, 1″-HCHand 1′-CH2); 13C NMR (CDCl3) δ: 36.1, 36.6, 55.3, 71.7, 72.1, 103.3, 113.9, 114.0, 127.0, 127.1, 135.1, 135.4, 159.2, 159.4, 161.4, 169.9.
3,5-bis[2-(3,4-dimethoxyphenyl)-2-hydroxyethyl]isoxazole (3c)
Yield: 1.12 g (65%); yellow crystals; mp 89-91 °C; IR (thin film) nmax:3384, 2937, 1599, 1513, 1420, 1260, 1138, 1022, 811, 762 cm-1; 1H NMR (CDCl3, 400 MHz) d: 6.89 (1H, s, ArH), 6.85 (1H, s, ArH), 6.84-6.74 (4H, m, ArH), 5.90 and 5.87 (1H, 2s, =CH), 4.93-4.81 (2H, m, 2″- and 2′-OCH), 3.81 and 3.80 (12H, 2s, 4´OCH3), 3.08 (1H, dd, J = 15.2 and 8.7 Hz, 1″-HCH), 3.03-2.86 (3H, m, 1″-HCHand 1′-CH2);13C NMR (CDCl3) δ: 36.0, 36.8, 55.8, 55.9, 71.7, 72.3, 103.3, 108.9, 109.1, 111.0, 117.9, 118.0, 135.8, 136.0, 148.4, 148.5, 148.9, 161.4, 169.9.
3-[2-phenyl-2-hydroxyethyl]-5-(2-(4-methoxyphenyl)-2-hydroxyethyl)isoxazole (3d)
Yield: 0.77 g (57%); brown crystals; mp 63-64°C; IR (thin film) nmax: 3365, 2896, 1606, 1513, 1437, 1246, 1176, 833, 701 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.34-7.20 (5H, m, ArH), 7.16 (2H, d, J = 8.7 Hz, ArH), 6.80 (2H, d, J = 8.7 Hz, ArH), 5.85 and 5.84 (1H, 2s, =CH), 4.91-4.80 (2H, m, 2″-and2′-OCH), 3.72 (3H, s, OCH3), 3.03 (1H, dd,J = 15.2 and 8.6 Hz, 1″-HCH), 2.99-2.83 (3H, m, 1″-HCHand 1′-CH2); 13C NMR (CDCl3) δ: 36.2, 36.6, 55.3, 71.8, 72.5, 103.3, 114.0, 125.8, 127.0, 127.8, 128.5, 135.0, 143.2, 159.4, 161.3, 169.9.
3-[2-phenyl-2-hydroxyethyl]-5-(2-(3,4-dimethoxyphenyl)-2-hydroxyethyl)isoxazole(3e)
Yield: 0.75 g (51%); yellow oil; IR (thin film) nmax: 3381, 2938, 1600, 1514, 1421, 1260, 1138, 1022, 810, 760, 701 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.33-7.19 (5H, m, ArH), 6.84 (1H, s, ArH), 6.80 (1H, d, J = 8.5 Hz, ArH), 6.76 (1H, d, J = 8.2 Hz, ArH), 5.87 and 5.85 (1H, 2s, =CH), 4.92-4.84 (2H, m, 2″- and 2′-OCH), 3.80 and 3.79 (6H, 2s, 2´OCH3) 3.07 (1H, dd, J = 15.1, 8.7 Hz, 1″-HCH), 3.02-2.86 (3H, m,1″-HCHand 1′-CH2);13C NMR (CDCl3) δ: 35.9, 36.7, 55.8, 71.6, 72.4, 103.2, 108.9, 111.0, 117.9, 125.7, 127.7, 128.4, 135.7, 143.2, 148.5, 148.9, 161.3, 169.9.
3-[2-(4-methoxyphenyl)-2-hydroxyethyl]-5-(2-(3,4-dimethoxyphenyl)-2-hydroxyethyl)isoxazole(3f)
Yield: 0.74 g (46%); green crystals;mp 76-77°C; IR (thin film) nmax: 3383, 2935, 1604, 1513, 1421, 1238, 1139, 1024, 811, 763 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.21 (2H, d, J = 8.2 Hz, ArH), 6.85 (1H, s, ArH), 6.84-6.79 (3H, m,ArH), 6.77 (1H, d, J = 8.2 Hz, ArH), 5.89 and 5.86 (1H, 2s, =CH), 4.93-4.80 (2H, m, 2″- and 2′-OCH), 3.82 and 3.81 (6H, 2s, 2´OCH3), 3.75 (3H, s, OCH3), 3.08 (1H, dd, J = 15.2 and 8.7 Hz, 1″-HCH), 3.03-2.85(3H, m, 1″-HCHand 1′-CH2);13C NMR (CDCl3) δ: 35.9, 36.7, 55.2, 55.8, 55.9, 71.7, 72.1, 103.2, 108.9, 111.0, 113.8, 117.9, 127.0, 135.4, 135.8, 148.5, 149.0, 159.1, 161.4, 169.9.
General procedure fordehydration of 3,5-bis(2-aryl-2-hydroxyethyl)isoxazoles3a-f
A mixture of 3,5-bis(2-aryl-2-hydroxyethyl)isoxazole(1.0 mmol), P2O5 (0.4mmol) in benzene (5 ml) was heated under reflux temperature for 2 hours. The benzene layer was decanted and the residue was washed once with hot benzene. The combined benzene layer was washed with water, dried (Na2SO4) and concentrated under reduced pressure. The residue was purified using column chromatography on silica gel with CH2Cl2 as eluent to give 5-(2-arylethenyl)-3-methylisoxazoles 2a-f(52-66%) as crystalline solids.
3,5-bis(2-phenylethenyl)isoxazole(2a)
Yield: 0.18 g (66%); white crystals; mp 186-187°C; IR (thin film) nmax:3033, 1644, 1579, 1559, 1429, 1292, 960, 751, 693 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.56-7.48(4H, m, ArH), 7.42-7.29 (7H, m, ArH and Ar1CH=CH-), 7.19 (1H, d, J = 16.6 Hz, Ar2CH=CH-), 7.13 (1H, d, J = 16.5 Hz, Ar2CH=CH-), 6.96 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 6.49 (1H, s, =CH); 13C NMR (CDCl3)δ: 98.5, 113.0, 116.1, 127.0, 127.1, 128.9, 129.2, 134.9, 135.5, 135.8, 162.0, 168.3.
3,5-bis(2-(4-methoxyphenyl)ethenyl)isoxazole(2b)
Yield: 0.20 g (61%); yellow crystals; mp 194-196 °C (lit.11 180-181 °C); IR (thin film) nmax: 2934, 1644, 1604, 1577, 1512, 1431, 1257, 1175, 1029, 967, 823, 753 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.48 (4H, d, J = 8.7 Hz, ArH), 7.31 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 7.13 (1H, d, J = 16.5 Hz, Ar2CH=CH-), 7.00 (1H, d, J = 16.5 Hz, Ar2CH=CH-), 6.92 (4H, d, J = 8.5 Hz, ArH), 6.83 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 6.43 (1H, s, =CH), 3.84 (6H, s, 2´OCH3);13C NMR (CDCl3) δ:55.4, 97.7, 111.0 114.0, 114.3, 128.3, 128.6, 128.7, 134.4, 135.2, 160.2, 160.5, 162.2, 168.5.
3,5-bis[2-(3,4-dimethoxyphenyl)ethenyl]isoxazole(2c)
Yield: 0.22 g (56%); white crystals; mp 169-171 °C (lit.11 159-160 °C); IR (thin film) nmax:2937, 1644, 1584, 1561, 1512, 1428, 1264, 1139, 1024, 963, 805, 735 cm-1;1H NMR (CDCl3, 400 MHz) d:7.22 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 7.09-6.93 (6H, m, ArH, Ar2CH=CH-andAr2CH=CH-),6.83-6.73 (3H, m, ArHand Ar1CH=CH-), 6.36 (1H, s, =CH), 3.90 and 3.89 (6H, 2s, 2´OCH3), 3.86 and 3.85 (6H, 2s, 2´OCH3);13C NMR (CDCl3) δ: 55.8, 97.8, 108.8, 109.1, 110.0, 111.1, 113.9, 120.9, 121.0, 128.5, 128.8, 134.5, 135.4, 149.1, 149.8, 150.1, 162.1, 168.4.
5-(2-(4-methoxyphenyl)ethenyl)-3-[2-phenylethenyl]isoxazole (2d)
Yield: 0.18 g (60%); yellow crystals; mp 155-156°C; IR (thin film) nmax:3035, 2928, 1643, 1603, 1509, 1430, 1254, 1173, 1032, 963, 822, 754, 695 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.53 (2H, d, J = 7.2 Hz, ArH), 7.47 (2H, d, J = 8.7 Hz, ArH), 7.42-7.28 (4H, m, ArHand Ar1CH=CH-), 7.18 (1H, d, J = 16.6 Hz, Ar2CH=CH-), 7.13 (1H, d, J = 16.5 Hz, Ar2CH=CH-), 6.92 (2H, d, J = 8.7 Hz, ArH), 6.83 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 6.44 (1H, s, =CH), 3.84 (3H, s, OCH3);13C NMR (CDCl3) δ: 55.4, 97.7, 110.9, 114.3, 116.2, 127.0, 128.3, 128.6, 128.8, 135.5, 135.6, 135.9, 160.5, 162.0, 168.7.
5-(2-(3,4-dimethoxyphenyl)ethenyl)-3-[2-phenylethenyl]isoxazole(2e)
Yield: 0.19 g (58%); white crystals; mp 153-154 °C; IR (thin film) nmax:2936, 1644, 1583, 1561, 1513, 1433, 1269, 1140, 1025, 964, 754, 697 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.47 (2H, d, J = 7.5 Hz, ArH), 7.39-7.28 (3H, m, ArH), 7.25 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 7.13 (1H, d, J = 16.6 Hz, Ar2CH=CH-), 7.08 (1H, d, J = 16.6 Hz, Ar2CH=CH-), 7.05-6.98 (2H, m, ArH), 6.78 (2H, m, ArHand Ar1CH=CH-), 6.39 (1H, s, =CH), 3.90 (3H, s, OCH3), 3.85 (3H, s, OCH3);13C NMR (CDCl3) δ: 55.8, 55.9, 97.9, 109.1, 111.0, 111.2, 116.1, 121.1, 126.9, 128.5, 128.8, 134.7, 135.6, 135.8, 149.2, 150.1, 161.9, 168.5.
5-(2-(3,4-dimethoxyphenyl)ethenyl)-3-[2-(4-methoxyphenyl)ethenyl]isoxazole(2f)
Yield: 0.19 g (52%); white crystals; mp 169-171 °C; IR (thin film) nmax: 2962, 1643, 1603, 1581, 1512, 1430, 1264, 1174, 1140, 1026, 965, 821, 750 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.45 (2H, d, J = 8.6 Hz, ArH), 7.27 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 7.10(1H, d, J = 16.6 Hz, Ar2CH=CH-), 7.05 (2H, d, J = 7.7 Hz, ArH), 6.98 (1H, d, J = 16.4 Hz, Ar2CH=CH-), 6.93-6.84 (3H, m,ArH), 6.81 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 6.40 (1H, s, =CH), 3.93(3H, s, OCH3), 3.90 (3H, s, OCH3), 3.82 (3H, s, OCH3);13C NMR (CDCl3) δ: 55.3, 55.9, 97.8, 109.1, 111.1, 111.2, 113.9, 114.3, 121.1, 128.3, 128.6, 134.6, 135.2, 149.2, 150.2, 160.2, 162.2, 168.4.
General procedure forreductive ring opening of 3,5-bis(2-arylethenyl)isoxazoles2a-f
A solution of a 3,5-bis(2-arylethenyl)isoxazole (0.4mmol), water (0.4mmol) and [Mo(CO)6] (0.2mmol) in acetonitrile (8 ml) was heated under refluxtemperaturefor 24 hours and cooled to room temperature. The reaction mixture was filtered through Celite and evaporated to dryness under reduced pressure. Purification ofthe residue using column chromatography on silica gel with 30% ethyl acetate in hexane as an eluent gave1,7-diaryl-5-amino-1,4,6-heptatrien-3-ones 8a-f(43-56%) as yellow crystalline solids or as yellow oils.
5-amino-1,7-diphenylhepta-1,4,6-trien-3-one (8a)
Yield: 61.5 mg (56%); yellow crystals; mp 145-147°C; IR (thin film) nmax: 3388, 3196, 1634, 1570, 1519, 1149, 966, 754, 694 cm-1; 1H NMR (CDCl3, 400 MHz) d:d7.60-7.54 (3H, m, ArH and Ar1CH=CH-), 7.49 (2H, d, J = 7.1 Hz, ArH) 7.41-7.30 (6H, m, ArH), 7.11 (1H, d, J = 16.4 Hz, Ar2CH=CH-), 6.79 (1H, d, J = 15.9 Hz, Ar2CH=CH-), 6.53 (1H, d, J = 16.4 Hz, Ar1CH=CH-), 5.53 (1H, s, =CHCO); 13C NMR (CDCl3) δ: 98.7, 124.8, 127.4, 127.9, 128.6, 128.8, 129.0, 129.4, 129.5, 134.5,135.2, 135.7, 138.7, 157.7, 188.4.
5-amino-1,7-bis(4-methoxyphenyl)-1,4,6-heptatrien-3-one(8b)
Yield: 66.8 mg (50%); yellow crystals; mp 158-160°C; IR (thin film) nmax: 3393, 3006, 1630, 1601, 1565, 1515, 1256, 1174, 754 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.56-7.47 (3H, m, ArH and Ar1CH=CH-), 7.43 (2H, d, J = 8.5 Hz, ArH), 7.05 (1H, d, J = 16.3 Hz, Ar2CH=CH-), 6.93-6.86 (4H, m, ArH), 6.66 (1H, d, J = 15.8 Hz, Ar2CH=CH-), 6.39 (1H, d, J = 16.3 Hz, Ar1CH=CH-), 5.47 (1H, s, =CHCO), 3.83 (6H, s, 2´OCH3);13C NMR (CDCl3)δ: 55.3, 55.4, 98.4, 114.2, 114.4, 122.4, 126.6, 128.0, 128.5, 128.8, 129.4, 134.0, 138.2, 158.1, 160.7, 188.3.
5-amino-1,7-bis(3,4-dimethoxyphenyl)-1,4,6-heptatrien-3-one(8c)
Yield: 76.2mg (48%); yellow oil; IR (thin film) nmax: 3417, 2937, 1629, 1587, 1513, 1461, 1263, 1139, 1024, 758 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.51 (1H, d, J = 15.7 Hz, Ar1CH=CH-), 7.14-7.01 (5H, m, ArH and Ar2CH=CH-), 6.85 (2H, d, J = 8.3 Hz, ArH),6.67 (1H, d, J = 15.8 Hz, Ar2CH=CH-), 6.39 (1H, d, J = 16.3 Hz, Ar1CH=CH-), 5.52 (1H, s, =CHCO),3.92and 3.90 (12H, 2s, 4´OCH3);13C NMR (CDCl3) δ: 55.9, 56.0, 98.3, 109.3, 109.7, 111.1, 111.2, 121.5, 122.2, 122.6, 126.7, 128.3, 128.8, 134.4, 138.5, 149.1, 149.3, 150.4, 158.1, 188.2.
5-amino-1-(4-methoxyphenyl)-7-phenyl-1,4,6-heptatrien-3-one (8d)
Yield: 66.0mg (54%); yellow crystals;mp 161-162°C; IR (thin film) nmax: 3364, 3008, 1631, 1602, 1564, 1515, 1256, 1145, 752 cm-1; 1H NMR ((CDCl3, 400 MHz) d: 7.56-7.45 (5H, m, ArH and Ar1CH=CH-), 7.42-7.29 (3H, m, ArH), 7.09 (1H, d, J = 16.4 Hz, Ar2CH=CH-), 6.88 (2H, d, J = 8.4 Hz, ArH), 6.67 (1H, d, J = 15.8 Hz, Ar2CH=CH-), 6.51 (1H, d,J = 16.4 Hz, Ar1CH=CH-), 5.49 (1H, s, =CHCO), 3.81 (3H, s, OCH3); 13C NMR (CDCl3)δ: 55.3, 98.8, 114.2, 124.9, 126.5, 127.3, 128.4, 128.9, 129.3, 129.5, 134.3, 135.3, 138.5, 157.5, 160.8, 188.6.
5-amino-1-(3,4-dimethoxyphenyl)-7-phenyl-1,4,6-heptatrien-3-one(8e)
Yield: 62.6mg (47%); yellow oil; IR (thin film) nmax: 3395, 2959, 1631, 1575, 1516, 1264, 1139, 756 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.55-7.47 (3H, m, ArH and Ar1CH=CH-), 7.41-7.31 (3H, m, ArH), 7.14-7.08 (3H, m, ArH and Ar2CH=CH-), 6.85 (1H, d, J = 8.3 Hz, ArH), 6.68 (1H, d, J = 15.8 Hz, Ar2CH=CH-), 6.52 (2H, d, J = 16.4 Hz, Ar1CH=CH-), 5.53 (1H, s, =CHCO),3.92(3H, s, OCH3), 3.90 (3H, s, OCH3); 13C NMR (CDCl3) δ: 55.9, 56.0, 98.6, 109.8, 111.2, 122.2, 124.9, 126.7, 127.3, 128.7, 128.9, 129.4, 134.3, 135.3, 138.7, 149.2, 150.5, 157.5, 188.5.
5-amino-1-(3,4-dimethoxyphenyl)-7-(4-methoxyphenyl)-1,4,6-heptatrien-3-one (8f)
Yield: 62.7 mg (43%); yellow oil; IR (thin film) nmax: 3394, 2964, 1597, 1514, 1262, 1141, 1027, 755 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.51 (1H, d, J = 15.7 Hz, Ar1CH=CH-), 7.43 (2H, d, J = 8.6 Hz, ArH), 7.15-7.02 (3H, m, ArH and Ar2CH=CH-), 6.92-6.82 (3H, m,ArH), 6.67 (1H, d, J = 15.7 Hz, Ar2CH=CH-), 6.39 (1H, d, J = 16.3 Hz, Ar1CH=CH-), 5.50 (1H, s, =CHCO), 3.92(3H, s, OCH3), 3.90 (3H, s, OCH3), 3.83 (3H, s, OCH3);13C NMR (CDCl3) δ:55.3, 55.9, 98.1, 109.7, 111.1, 114.4, 122.2, 122.3, 126.9, 128.0, 128.8, 134.2, 138.4, 149.1, 150.4, 158.3, 160.7, 188.1.
General procedure forhydrolysis of 5-amino-1,7-diaryl-1,4,6-heptatrien-3-ones 8a-f
To a stirred solution of 5-amino-1-aryl-1,4-hexadien-3-one (0.1mmol) in ethanol (1.5 ml)was added concentrated HCldropwise to adjust the pH between 4 and 5. The solution was then stirred at 50°C. After 24 hours,the reaction mixture was neutralized with a saturated K2CO3 solution.The aqueous layer was extracted with diethyl ether (3 × 25 mL). The combined organic layer was dried (Na2SO4) and concentrated under reduce pressure. Purification of the residue using column chromatography on silica gel with a gradient of 30-50% ethyl acetate in hexane as eluent gave1-aryl-5-hydroxy-1,4-hexadien-3-ones1a-f (48-63%) as crystalline solids.
5-Hydroxy-1,7-diphenylhepta-1,4,6-trien-3-one (1a)
Yield: 14.4mg (52%); yellow crystals; mp 146-148°C; IR (thin film) nmax: 3057, 1627, 1581, 1445, 1275, 1139, 972, 753 cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.67 (2H, d, J = 15.9 Hz, Ar1CH=CH- and Ar2CH=CH-), 7.56 (4H, dd, J = 7.4 and 1.8 Hz, ArH), 7.44-7.34 (6H, m, ArH), 6.64 (2H, d, J = 15.9 Hz, Ar2CH=CH– and Ar1CH=CH-), 5.86 (1H, s, =CHCO); 13C NMR (CDCl3) δ: 101.8, 124.1, 128.1, 128.9, 130.1, 135.0, 140.6, 183.3.
5-Hydroxy-1,7-bis(4-methoxyphenyl)-1,4,6-heptatrien-3-one(1b)
Yield: 21.2 mg (63%); orange crystals;mp 175-177°C; IR (thin film) nmax: 3037, 2933, 2838, 1628, 1600, 1574, 1512, 1461, 1253, 1173, 1138, 1031, 974, 828cm-1; 1H NMR (CDCl3, 400 MHz) d: 7.62 (2H, d, J = 15.8 Hz, Ar1CH=CH- and Ar2CH=CH-), 7.50 (4H, d, J = 8.7 Hz,ArH), 6.91 (4H, d, J = 8.7 Hz, ArH), 6.49 (2H, d, J = 15.8 Hz, Ar2CH=CH– and Ar1CH=CH-), 5.78 (1H, s, =CHCO), 3.84 (6H, s, 2´OCH3); 13C NMR (CDCl3) δ: 55.4, 101.3, 114.4, 121.8, 127.8, 129.8, 140.1, 161.3, 183.3.
5-Hydroxy-1,7-bis(3,4-dimethoxyphenyl)-1,4,6-heptatrien-3-one(1c)
Yield: 19.5 mg (49%); orange crystals; mp115-117°C; IR (thin film) nmax:2935, 2837, 1624, 1583, 1512, 1462, 1262, 1136, 1023, 969, 808 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.61 (2H, d, J = 15.8 Hz, Ar1CH=CH- and Ar2CH=CH-), 7.14 (2H, d, J = 8.3 Hz, ArH), 7.08 (2H, s, ArH), 6.88 (2H, d, J = 8.3 Hz, ArH), 6.50 (2H, d, J = 15.8 Hz, Ar2CH=CH– and Ar1CH=CH-), 5.82 (1H, s, =CH), 3.93(6H, s, 2´OCH3), 3.92 (6H, s, 2´OCH3); 13C NMR (CDCl3) δ: 55.9, 56.0, 101.3, 109.8, 111.2, 122.1, 122.6, 128.1, 140.4, 149.3, 151.1, 183.3.
5-Hydroxy-1-(4-methoxyphenyl)-7-phenyl-1,4,6-heptatrien-3-one (1d)
Yield: 14.7 mg (48%); yellow crystals; mp 140-142°C; IR (thin film) nmax: 3050, 2933, 2836, 1623, 1601, 1576, 1512, 1447, 1255, 1175, 1140, 1028, 977, 826 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.64 (1H, d, J = 15.9 Hz, Ar2CH=CH-), 7.63 (1H, d, J = 15.8 Hz, Ar1CH=CH-), 7.55 (2H, d, J = 7.5 Hz, ArH), 7.51 (2H, d, J = 8.7 Hz, ArH), 7.43-7.35 (3H, m, ArH), 6.91 (2H, d, J = 8.2 Hz, ArH), 6.61 (1H, d, J = 15.9 Hz, Ar2CH=CH-), 6.51 (1H, d, J = 15.8 Hz, Ar1CH=CH-), 5.81 (1H, s, =CHCO), 3.83 (3H, s, OCH3); 13C NMR (CDCl3) δ: 55.4, 101.6, 114.4, 121.8, 124.1, 127.7, 128.1, 128.9, 129.8, 135.1, 140.1, 140.6, 161.4, 182.2, 184.4.
5-Hydroxy-1-(3,4-dimethoxyphenyl)-7-phenyl-1,4,6-heptatrien-3-one (1e)
Yield: 17.1 mg (51%); orange crystals;mp 138-139°C; IR (thin film) nmax:2934, 2837, 1624, 1581, 1511, 1446, 1420, 1261, 1136, 1023, 968 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.65 (1H, d, J = 15.9 Hz, Ar2CH=CH-), 7.62 (1H, d, J = 15.7 Hz, Ar1CH=CH-), 7.55 (2H, d, J = 7.6, ArH), 7.43-7.34 (3H, m, ArH), 7.15 (1H, d, J = 8.3 Hz, ArH), 7.08 (1H, s,ArH), 6.88 (1H, d, J = 8.3 Hz, ArH), 6.62 (1H, d, J = 15.8 Hz, Ar2CH=CH-), 6.52 (1H, d, J = 15.8 Hz, Ar1CH=CH-), 5.84 (1H, s, =CHCO), 3.94(3H, s, OCH3), 3.92 (3H, s, OCH3);13C NMR (CDCl3) δ: 55.9, 56.0, 101.5, 109.9, 111.2, 122.1, 122.8, 124.1, 128.0, 128.1, 128.9, 130.0, 135.1, 140.2, 140.8, 149.3, 151.2, 182.4, 184.2.
5-Hydroxy-1-(3,4-dimethoxyphenyl)-7-(4-methoxyphenyl)-1,4,6-heptatrien-3-one(1f)
Yield: 19.0 mg (52%); orange crystals;mp 155-157°C; IR (thin film) nmax: 3002, 2935, 1625, 1595, 1512, 1461, 1256, 1172, 1136, 1025, 970, 829 cm-1; 1H NMR (CDCl3, 400 MHz) d:7.62 (1H, d, J = 15.8 Hz, Ar2CH=CH-), 7.60 (1H, d, J = 15.8 Hz, Ar1CH=CH-), 7.50 (2H, d, J = 8.7 Hz, ArH), 7.13 (1H, d, J = 8.3 Hz, ArH), 7.08 (1H, d, J = 1.5 Hz, ArH), 6.91 (2H, d, J = 8.7 Hz, ArH),6.87 (1H, d, J = 8.3 Hz, ArH), 6.49 (2H, d, J = 15.8 Hz, Ar1CH=CH– and Ar2CH=CH-), 5.80 (1H, s, =CHCO), 3.93(3H, s, OCH3), 3.91 (3H, s, OCH3), 3.84 (3H, s, OCH3); 13C NMR (CDCl3) δ: 55.4, 55.9, 56.0, 101.3, 109.8, 111.2, 114.4, 121.8, 122.1, 122.6, 127.8, 128.1, 129.8, 140.2, 140.3, 149.3, 151.1, 161.3, 183.1, 183.4.
Results and Discussion
3,5-Dimethylisoxazole (5) used in our study was readily prepared in 82% yield from condensation between 2,4-pentanedione (6) with hydroxylamine hydrochloride in aqueous ethanol at reflux temperature for 3 hours.10
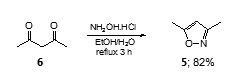
Lateral metalation of 3,5-dimethylisoxazole (5) occurred regiospecificallyat the C5-methyl group upon treatment with eithern-BuLi12 or NaNH2.13Metalation at the 3-methyl group could be accomplished when metalating agent was eithers-BuLi or t-BuLi.14
As expected, lateral metalation of 3,5-dimethylisoxazole (5) with n-BuLi in THF at -78°C for 1 hour followed by quenching with aromatic aldehydes7a-c gave the corresponding 5-(2-aryl-2-hydroxyethyl)-3-methylisoxazoles 4a-c in good yields (Scheme 2).
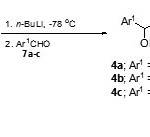 |
Scheme 2 Click here to View scheme |
Further treatment of 5-(2-aryl-2-hydroxyethyl)-3-methylisoxazoles 4a-c with 2 equivalents of s-BuLi at -78°Cfollowed by quenching with aromatic aldehydes 7a-c led to the formation of four diastereomers ofthe corresponding 3,5-bis(2-aryl-2-hydroxyethyl)isoxazoles3a-f in moderate yields (Scheme 3).
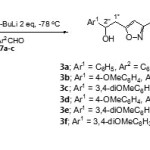 |
Scheme 3 Click here to View scheme |
Since P2O5 is a very efficient dehydrating agent, it was used as such in dehydration of 3,5-bis(2-aryl-2-hydroxyethyl)isoxazoles3a-f.In benzene at reflux temperature15 (Scheme 4), the reaction between carbinols3a-f and P2O5 furnished the corresponding 3,5-bis(2-arylethenyl)isoxazoles2a-f in moderate yields.
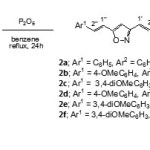 |
Scheme 4 Click here to View scheme |
The weak N–O bond ofisoxazole ring is easily cleaved into b-aminoenonebyeithercatalytic hydrogenolysis with platinum or palladium and Raney nickel under normal pressure and temperature16,17orby treatment with transition metal carbonyls such as molybdenum hexacarbonyl [Mo(CO)6].18To avoid catalytic hydrogenation at the two styryl groups, 3,5-bisstyrylisoxazoles2a-fwere treated with molybdenum hexacarbonyl in moist acetonitrile at reflux temperature. This treatment successfully provided the corresponding1,7-diaryl-5-amino-1,4,6-heptatrien-3-ones 8a-f in moderate yields ( Scheme 5).
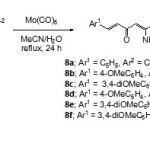 |
Scheme 5 Click here to View scheme |
b-Aminoenones are easily transformed into b-hydroxyenones by simple acidic hydrolysis.19,20 In our final step, hydrolysis of b-aminoenones8a-f was carried out by treatment with hydrochloric acid in ethanol at pH 4-5. The corresponding curcumin derivatives, in their enol forms 1a-f were obtained in satisfied yields (Scheme 6).
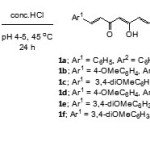 |
Scheme 6 Click here to View scheme |
Conclusions
The present six steps procedure provides a novel alternative route to symmetrical and unsymmetrical curcumin derivatives. Good to moderate yields of expected products were obtained from each step.
Acknowledgement
We are pleased to acknowledge financial support from the Center for Innovation in Chemistry (PERCH-CIC) and the Commission of Higher Education (CHE-RG), Ministry of Education.
References
- Anand P, Thomas SG, Kunnumakkara AB, Sundaram C, Harikumar KB, Sung B, et al. Biological activities of curcumin and its analogues (Congeners) madeby man and Mother Nature.BiochemPharmacol.(2008),76, 1590-1611.
CrossRef - Ferrari E,Pignedoli F, Imbriano C, Marverti G, Basile V, Venturi E, et al. Newly synthesized curcumin derivatives: Crosstalk between chemico-physical properties and biological activity. J Med Chem.(2011), 54, 8066-77.
CrossRef - Weber WM, Hunsaker LA, Abcouwer SF, Deck LM, Jagt DLV Anti-oxidant activaties ofcurcumin and relatedenones.Bioorg Med Chem.(2005),13, 3811-20.
CrossRef - Kim HK, Yang CH Synthetic curcumin derivatives Inhibit Jun-Fos-DNA complex formation. Bull KoreanChemSoc.(2004), 25, 1769-74.
CrossRef - Nurfina AN, Reksohadiprodjo MS, Timmerman H, Jenie UA, Sugiyanto D, van der Goot H Synthesis of some symmetrical curcumin derivatives and their anti-inflammatory activity.Eur J Med Chem.(1997), 32, 321-8.
CrossRef - Pabon HJJ A synthesis of curcumin and related compounds.Rec TrawChim.(1964),83, 379-86.
CrossRef - Shao WY, Cao YN, Yu ZW, Pan WJ, Qiu X, Bu XZ, An LK, Huang ZS, Gu LQ, Chan LSC Facile preparation of new unsymmetrical curcuminderivatives by solid-phase synthesis strategy, Tetrahedron Letters. (2006), 47, 4085-9.
CrossRef - Leonard J, Lygo B, Procter G Advanced Practical Organic Chemistry2nd edn.
- Blackie Academic and Professional,Glasgow,(1995),100-1.
- Kofron WG, Baclawski LM A convenient method for estimation of
CrossRef - alkyllithium concentrations.JOrg Chem.(1976),41, 1879-80.
- Fitton AO, Smalley RK Practical Heterocyclic Chemistry. Academic press;London, (1968), 28.
- Changtam C, Hongmanee P, Suksamrarn A Isoxazole analogs ofcurcuminoids with highly potent multidrug-resistant antimycobacterial activity.EuropeanJournal of Medicinal Chemistry.(2010),45, 4446-57.
CrossRef - Micetich RG Lithiation of five-membered heteroaromatic compounds. The methyl substituted 1,2-azoles, oxadiazoles, and thiadiazoles.Can JChem.(1970),48, 2006-14.
CrossRef - Kashima C, Tobe S, Sugiyama N, Yamamoto M The alkylation of 3,5-dimethyl isoxazole. Bull ChemSoc Japan.(1973),46,310-3.
CrossRef - Brunelle DJ Isoxazoles as β-diketonesynthons-selective anion formation on 3,5-dialkylisoxazoles. Tetrahedron Lett.(1981),22, 3699-702.
CrossRef - Cermak J, Nguyen Thi TH, Veelak, Krupkova A Dehydration of (perfluoroalkyl)tetramethylcyclopentenols. Molecules.(2011),16, 4031-44.
CrossRef - BaraldiPG, Barco A , BenettiS, MoroderF, PolliniPG, Simoni D 3,5-Disubstituted isoxazoles as synthon for (±)-pyrenophorin and (±)-vermiculine synthesis. J Org Chem. (1983), 48, 1297-302.
CrossRef - Akhrem AA, Lakhvich FA, KhripachVA,Klebanovich IB Nitrile in a new approach to the synthesis of glutarimide antibiotics.Tetrahedron Lett.(1976),17,3983-84.
CrossRef - Nitta M, Kobayashi T Reductive ring opening of isoxazoles with Mo(CO)6 and water.JChemSoc,ChemCommun.(1982),20,1401-6.
CrossRef - Kenar JA Preparation of long-chain β-enaminones and β-diketones from long- chain 3,5-disubstituted isoxazole compounds.J Am Oil ChemSoc.(2003),80,1027-32.
CrossRef - Akhrem AA, Lakhvich FA, Khripach VA, Klebanovich IB Synthesis of glutarimide antibiotics and their analogs through 1,3-cycloaddition of3-glutarimidylacetonitrile oxide to 2-cyclohexenones.ChemHeterocyclCompd.(1979),15, 194-8.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


