Covalently supportedSulfonic and acetic acids ontopolypyrrole asgreen, cheap and recoverablesolid acid catalysts for the synthesis of 4H-pyrano[2,3-c]pyrazoles
Elham Fadovipoor1,*,Simin Nazari1, Amanollah Zarei Ahmady2, Maryam Gorjizadeh3, Mozhgan Afshari3,Mosadegh Keshavarz4,*
1Department of Chemistry, Sousangerd Branch, Islamic Azad University, Sousangerd 44181-6189, Iran.
2Department of Medicinal Chemistry, School of Pharmacy,Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran.
3Department of Chemistry, Shoushtar Branch, Islamic Azad University, Shoushtar, Iran.
4Department of Gas and Petroleum, Yasouj University, Gachsaran, Iran.
Corresponding author: E-mail address: chem.mosadegh@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/310215
Article Received on :
Article Accepted on :
Article Published : 25 Jun 2015
Sulfonatedpolypyrrolenanospheres and polypyrrole N-functionalized acetic acid were synthesized. The catalytic efficiency of the prepared solid acid catalysts for the synthesis of 4H-pyrano[2,3-c]pyrazolesderivatives was evaluated and compared. The results showed that, regarding the time and yield of the products, the sulfonatedpolypyrrolenanospheres catalyst has better catalytic activity than polypyrrole N-functionalized acetic acid during first andsecond runs but shows a slight decline in its activityduring thethird run.
KEYWORDS:Polypyrrole N-functionalized acetic acid; Sulfonatedpolypyrrolenanospheres; Nano; solid catalyst; Heterogeneous; 4H-pyrano[2,3-c]pyrazoles
Download this article as:| Copy the following to cite this article: Fadovipoor E, Nazari S, Ahmady A. Z, Gorjizadeh M, Afshari M, Keshavarz M. Covalently supportedSulfonic and acetic acids ontopolypyrrole asgreen, cheap and recoverablesolid acid catalysts for the synthesis of 4H-pyrano[2,3-c]pyrazoles. Orient J Chem 2015;31(2). |
| Copy the following to cite this URL: Fadovipoor E, Nazari S, Ahmady A. Z, Gorjizadeh M, Afshari M, Keshavarz M. Covalently supportedSulfonic and acetic acids ontopolypyrrole asgreen, cheap and recoverablesolid acid catalysts for the synthesis of 4H-pyrano[2,3-c]pyrazoles. Available from: http://www.orientjchem.org/?p=9447 |
Introduction
Heterogenization of homogeneous catalysts has been an interesting area of research from the industrial point of view; this combines the advantages of homogeneous catalysts (high activity and selectivity, etc.) with the engineering advantages of heterogeneous catalysts such as easy catalyst separation, long catalytic life, easy catalyst regenerability, thermal stability and recyclability.1 Several types of solid sulfonic acid functionalized silica (both amorphous and ordered) have been synthesized and applied as an alternative to traditional sulfonic acid resins and homogeneous acids in catalyzing chemical transformations.2-5From this viewpoint, catalytic reactions lead to valuable processes, since the use of stoichiometric reagents that are often toxic pose essential restrictions from both an economical and an environmental standpoint and have direct relation to product purification and waste controlling.6Use of solid acids in organic reactions has significant roles, because these species have many rewards such as ease in handling, reduced reactor and plant corrosion harms, and more environmentally benign disposal.7–12Green chemistry not only needs the use of environmentally benign reagents and solvents, but also the recovery and reuse of the catalyst. Fromthis perspective, some types of solid sulfonic acid functionalized silica have been synthesized and used as an alternate to traditional homogeneous acids in catalyzing chemical transformations.
Recently, nanotubular and nanosphereconducting polymers have attracted much attention due to their interesting electrochemical properties caused by their small dimensions and high surface area.13–16 Among the reported conducting nanopolymers, polypyrrole nanotubes (PPyNTs) and polypyrrolenanospheres (PPyNs) have received attractiveness because of redox properties, their relatively high conductivity, easy preparation, ion exchange capacity, excellent environmental stability.17-19Moreover, PPyNTs and PPyNs can be synthesized chemically in a bulk amount from benign aqueous environment in ambient conditions, without the use of high technology and sophisticated instruments.
Pyrano[2,3-c]pyrazoles establish important precursors to hopeful drugs in the field of medicinal chemistry and exhibit wide range of biological activities.20–22 Therefore, development and improvement of new synthetic approaches in this field using easily accessible and benign catalysts seems to be a remarkable challenge. These compounds were first obtained by H. Otto in 1974 by adding malononitrile to 4-arylidene-3-methyl-2-pyrazolin-5-one.23 Several methods have been described for the preparation of this class of compounds.24-35 However, many of these methods suffer from certain drawbacks such as low yield, harsh reaction conditions, prolonged reaction time, and application of hazardous and/or costly catalysts and solvents. Therefore, development of greener, clean, and environmentally friendly approaches is still in demand. Due to the importance of pyrano[2,3-c]pyrazoles compounds, herein, we wish to introduce sulfonatedpolypyrrolenanospheres and polypyrrole N-functionalized acetic acid as new efficient and recyclable catalysts for one-pot three-component synthesis of highly functionalized 4H-pyrano[2,3-c]pyrazoles1a–kfrom the reactions between aldehydes, malononitrile and 3-methyl-1-phenyl-2-pyrazolin-5-one.
Experimental
General
Products were characterized by comparison of their spectroscopic data (1HNMR, 13CNMR and IR) and physical properties with those reported in the literature. NMR spectra were recorded in DMSO-d6 or CDCl3 on Bruker Advanced DPX 400 MHz instrument spectrometers using TMS as internal standard. IR spectra were recorded on a BOMEMMB-Series 1998 FT-IR spectrometer.ultrasonicator (Elma Transsonic T460/H) was used for ultrasonication. All yields refer to isolated products.
Preparation polypyrrolenanospheres (PPyNs)
In a typical procedure, in a round bottom flask containing 120 ml of deionized water, 4.0 g of decyl alcohol (1-decanol, 99%, Aldrich) was added and the mixture was stirring at room temperature for 40 min. Then, 6.0 g of dodecyltrimethylammonium bromide (DTAB, 99%, Aldrich) was added. After further stirring at 0 ◦C for 30 min, the emulsion was moved to an ultrasonicator (Elma Transsonic T460/H of capacity 1.5 liters and frequency of 35 kHz). Then, 3.2 g of pyrrole (98%, Aldrich) was added drop-wise to the mixture. To initiate polymerization, 8.0 g of FeCl3 (99%, Aldrich) was added. After ultrasonication for 3 h, the solid PPyNs were separated by filtration, washed with ethanol to remove the surfactant, and dried in an oven at 60 ◦C overnight.
Preparation of sulfonatedPolyperrolenanospheres (SPPyNs)
PyPNs (0.5 g) was heated in 100 ml of concentrated H2SO4 at 150 ◦C for 4 h. After cooling down to room temperature, ethanol (1000 ml) was added. The black solid was collected by filtration, and washed repeatedly using ethanol until no sulfate ions were detected in the filtrate.36
Preparation of Polyperrolenanotubes (PPyNTs)
The synthesis of PPyNTs was carried out by a self-degraded template method as reported in the literature.38In a typical procedure, 0.24 g (1.5 mmol) of FeCl3 was dissolved in 30 mL of 5 mM MO solution (0.15 mmol). Pyrrole monomers (105 mL, 1.5 mmol) were then added and the mixture was stirred at room temperature for 24 h. The formed PPy precipitate was washed with deionized water/ethanol several times until the filtrate was colourless and neutral. The produced PPyNTs was finally dried in dynamic vacuum at 60 ◦C for 24 h.
Preparation of Polyperrole nanotubes N-functionalized acetic acid (PPyNTsAA)
A mixture of PPyNTs (0.1 g), KOH powder (0.15 g), was dispersed in 25 mL DMF and sonicated for 1hand ethyl chloroacetate (0.61 g, 5 mmol) was added and then held at 60 ◦C for 12 h under vigorous agitation. Then HCl (20%) was added until the PH of the mixture decreased to 5 and heated again at 60 ◦C for another 2 h. Finally, the solid black precipitate was filtrated and washed with deionized water and ethanol to thoroughly remove physically absorbed HCl and unreacted ethyl chloroacetate from the surface of PPyNTsAA.
General procedure for synthesis of 4H-pyrano[2,3-c]pyrazolesderivatives
3-methyl-1-phenyl-2-pyrazolin-5-one (1 mmol), aldehyde (1 mmol) and malononitrile (1 mmol), were placed together in a round-bottom flask containing 5 mL of EtOH. SPPyNs (0.01 g) or PPyNTsAA (0.03 g) was added to the mixture and the suspension was magnetically stirred at reflux condition for appropriate time according to (Table 2). After completion of the reaction as followed by TLC (n-hexane: ethyl acetate; 3:1), the catalyst was filtered and washed with hot ethanol (2×5 mL). The recovered catalyst was washed with acetone, dried and stored for other similar consecutive runs. The filtrate mixture was recrystallized to provide the pure crystals of 4H-pyrano[2,3-c]pyrazole derivatives. The products are known compounds and are characterized by IR and NMR spectroscopy data for new compounds. Their melting points are compared with reported values. 24-35
Spectral data for selected products
6-Amino-4-(4-methoxyphenyl)-3-methyl-1-phenyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (1d)
Pale yellow powder; Yield: 0.32g (89%); mp 241-243 oC (242-243 oC). IR (KBr): = 3394, 3325, 3059, 2975, 2193, 1661, 1597, 1515, 1397, 1258 cm-1. 1H NMR: δ = 1.78 (3H, s, CH3), 3.74 (3H, s, OCH3), 4.62 (1H, s, CH), 6.89 (2H, d, J = 8.5 Hz, 2CH), 7.15 (2H, s, 2CH),7.17 (2H, s, NH2), 7.31 (1H, t, J = 7.6 Hz, CH), 7.49 (2H, t, J = 7.6 Hz, 2CH), 7.77 (2H, d, J = 8.5 Hz, 2CH) ppm. 13C NMR: δ = 12.6, 35.9, 55.1, 58.6, 98.9, 113.8, 119.9, 120.1, 126.2, 128.8, 129.4, 135.6, 137.5, 145.4, 158.2, 159.3 ppm.
6-Amino-4-(3-nitrophenyl)-3-methyl-1-phenyl-1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitrile (1e)
Yellow powder; Yield: 0.33g (88%); mp 190-192 oC (189-191 oC). IR (KBr): = 3436, 3296, 3098, 2190, 1651, 1589, 1517, 1446, 1386, 1349, 1258, 1119 cm-1. 1H NMR: δ = 1.90 (3H, s, CH3), 4.81 (1H, s, CH), 4.83 (2H, s, NH2), 7.36-7.37 (1H, m, CH), 7.48-7.51 (2H, m, 2CH), 7.56-7.59 (1H, m, CH), 7.66-7.67 (3H, m, 3CH), 8.13 (1H, s, CH), 8.19 (1H, d, 3JHH= 7.2 Hz, CH) ppm. 13C NMR: δ = 13.0, 36.8, 57.6, 98.0, 120.3, 120.5, 122.7, 126.7, 129.7, 130.7, 135.2, 137.9, 144.5, 145.6, 146.4, 184.4, 160.3 ppm.
Results and discussion
The proposed mechanism followed to obtain sulfonatedpolyperrolenanospheres (SPPyNs) and Polyperrole nanotubes N-functionalized acetic acid (PPyNTsAA) are outlined in scheme 1 and 2.
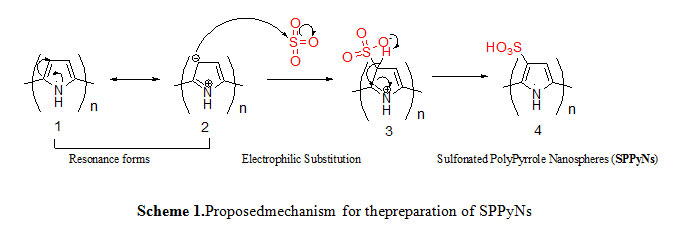 |
Scheme1: Proposedmechanism for thepreparation of SPPyNs Click here to View scheme |
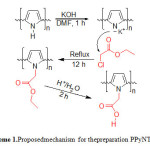 |
Scheme1A: Proposedmechanism for thepreparation PPyNTsAA Click here to View scheme |
Figure 1 shows the FESEM image of polypyrrolenanospheres (PPyNs). From figure 1, it can be seen that PPyNs have spherical shape with an average particle size of around 70-80 nm in diameter. The FESEM image of sulfonatedpolypyrrolenanospheres (PPyNs) shows that the particle surfaces are similar to that before sulfonation but the particles have become smaller in size with an average diameter of about 60-70 nm (Figure 2). The FTIR spectra of the SPPyNs exhibits a peak around 1550 cm−1 which is attributed to the C=C stretching vibration of pyrrole.The peaks at 1700 and 1300 cm−1 are assigned to C=N and C–N vibrations in PPy.A characteristic peak around 1045 cm−1 is attributed to the symmetric O=S=O stretching vibrations and confirms the presence of –SO3H groups in SPPyNs. Moreover, the peaks at around 800 and 650 cm−1 are assignable for S–O and C–S stretching vibrations, respectively.After the 3th reaction run, SPPyNs catalyst still possessed these peaks, indicating a good catalyst recyclability of SPPNs.
 |
Figure1: FESEM images of Polyperrolenanospheres (PPyNs) Click here to View figure |
 |
Figure2: FESEM images of sulfonatedPolyperrolenanospheres (SPPyNs) Click here to View figure |
In order to prepare PPyNTsAA catalyst, a mixture of PPyNTs and KOH powder was dispersed in DMF and sonicated to deprotonate PPyNTs. Then, ethyl chloroacetate was added and then heated at 60 ◦C for 12 h under vigorous agitation. Then aqueous HCl was added until the PH of the mixture decreased to 5 and heated again for further 2 h. Finally, the solid black precipitate was filtrated and washed with deionized water and ethanol to thoroughly remove physically absorbed HCl and unreacted ethyl chloroacetate from the surface of PPyNTsAA.
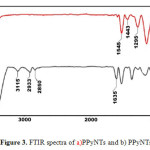 |
Figure3: FTIR spectra of a)PPyNTs and b) PPyNTsAA Click here to View figure |
Figure 3 shows the FTIR spectra of PPyNTs and PPyNTsAA. From figure 3b, two characteristic picks confirm the successful supporting of acetic acid on the surface of polypyrrole nanotubes. A newly observed characteristic pick at 1635 cm-1 is assigned to C=O stretching vibration of carboxylic group, and also a new broad peak between 3000-4000 cm-1 is attributed to OH of carboxylic acid.
On the base of our best knowledge, there is only one report that has investigated the catalytic activity of SPPyNs.36Moreover; this is the first report on the preparation of PPyNTsAA. Herein; we became interested in investigating the catalytic activity of the prepared SPPyNs and PPyNTsAA as new heterogeneous polymer based acid catalysts for the synthesis of 4H-pyrano[2,3-c]pyrazoles derivatives.
To optimize the reaction conditions, the reaction of the reaction of benzaldehyde, malononitrile and 3-methyl-1-phenyl-2-pyrazolin-5-one was selected as model to examine the effect of SPPyNs and PPyNTsA Acatalysts under a variety of conditions (Table 1).
The present optimization studies revealed that the best results were achieved by caring out the reaction in the presence of 0.01 g of SPPyNsand 0.03 g of PPyNTsAAunder reflux condition in ethanol (Table 1,entries 9 and 10). The larger amounts of the catalysts (0.015 g of SPPyNs and o.4 g ofPPyNTsAA) did not improve the yield while decreasing the amount of the catalyst led to decreased yield. Using these optimized conditions, the reaction of aromatic aldehydes, malononitrile and 3-methyl-1-phenyl-2-pyrazolin-5-one was explored (Scheme 3).
![Table 1.Investigation of catalytic activity of SPPyNs catalyst for the synthesis of 4H-pyrano[2,3-c]pyrazoles under various conditions](http://www.orientjchem.org/wp-content/uploads/2015/06/Vol31_No2_Cova_Elha_T1-150x150.jpg) |
Table1: Investigation of catalytic activity of SPPyNs catalyst for the synthesis of 4H-pyrano[2,3-c]pyrazoles under various conditions Click here to View table |
![Scheme 3.Synthesis of 4H-pyrano[2,3-c]pyrazolederivatives using SPPyNs and PPyNTsAA](http://www.orientjchem.org/wp-content/uploads/2015/06/Vol31_No2_Cova_Elha_Sch3-150x150.jpg) |
Scheme3: Synthesis of 4H-pyrano[2,3-c]pyrazolederivatives using SPPyNs and PPyNTsAA Click here to View scheme |
Table 2 represents the reaction times, Yields and the product of different synthesized 4H-pyrano[2,3-c]pyrazole derivatives from various substrates using SPPyNs and PPyNTsAA as catalysts. All the products were cleanly isolated with simple filtration and recrystallization from hot ethanol.In all the cases, aromatic aldehydes substituted with either electron-donating or electron-withdrawing groups smoothly underwent the reaction and gave the target products in good to excellent yields.
Table2: Synthesis of 4H-pyrano[2,3-c]pyrazole derivatives catalyzed by SPPyNs and PPyNTsAA
|
Product |
Ar |
SPPyNs Catalyst |
PPyNTsAA Catalyst |
||
|
Time(min) |
Yield (%)a |
Time(min) |
Yield(%)a |
||
|
1a |
C6H5 |
30 |
78 |
35 |
80 |
|
1b |
4-PhC6H4 |
40 |
81 |
50 |
79 |
|
1c |
4-MeC6H4 |
35 |
79 |
40 |
84 |
|
1d |
4-MeOC6H4 |
40 |
85 |
45 |
81 |
|
1e |
3-NO2C6H4 |
25 |
82 |
30 |
85 |
|
1f |
4-ClC6H4 |
30 |
81 |
45 |
89 |
|
1g |
2,4-(Cl)2C6H3 |
25 |
89 |
40 |
79 |
|
1h |
4-CNC6H4 |
30 |
86 |
50 |
88 |
|
1i |
4-OHC6H4 |
45 |
79 |
60 |
84 |
|
1j |
4-BrC6H4 |
40 |
80 |
50 |
87 |
|
1k |
2-OHC6H4 |
45 |
78 |
60 |
80 |
a Yield refer to isolated and pure product
In order to investigate the reusability of the prepared catalysts, the reaction of benzaldehyde, malononitrile and 3-methyl-1-phenyl-2-pyrazolin-5-one was selected again as the model reaction using SPPyNs and PPyNTsAA as catalysts.After completion of the reactions, the mixtures were filtered and the recovered catalysts were washed with acetone and dried before using for next consecutive runs (2 runs). The results showed that, regarding the time and yield of the products, the sulfonatedpolypyrrolenanospheres catalyst has better catalytic activity than polypyrrole N-functionalized acetic acid during first to third runs but shows a slight decline in its activity during the fourth and after runs.
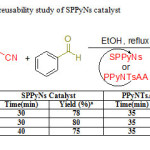 |
Table3: Recyclability and reusability study of SPPyNs catalyst Click here to View table |
Conclusion
In conclusion, SPPyNs and PPyNTsAA catalystswere simply prepared. The prepared heterogeneous catalysts were well characterized. The catalytic efficiency of the prepared catalysts was investigated for the preparation of 4H-pyrano[2,3-c]pyrazole derivatives. In the presence of the prepared solid acid catalysts, all the reactions were performed in short reaction times at high yields. It was shown that the title catalysts can be recovered and reused at least for three cycles.
Acknowledgement
We are grateful to the research council of Sousangerd Branch, Islamic Azad University and Yasouj University for financial support.
References
- Li Z, Ma X, Liu J,Feng X, Tian G, Zhu A,J. Mol. Catal. A: Chem.2007, 272, 132.
- Zareyee D,Karimi B,Tetrahedron Lett. 2007, 48, 1277.
- Niknam K, Saberi D,Tetrahedron Lett. 2009, 50, 5210.
- Niknam K, Saberi D, Sadegheyan M,Deris A,Tetrahedron Lett. 2010, 51, 692.
- Niknam K, Mohammadizadeh M R, Mirzaee S, SaberiD,Chin. J. Chem. 2010, 28, 663.
- FerreiraP,PhillipsE,RipponD,TsangSC,Appl. Catal. B,2005, 61, 206.
- KarimiB,KhalkhaliM,J. Mol. Catal. A: Chem.2005, 232, 113.
- MeleroJA,van GriekenR,MoralesG,Chem. Rev.2006, 106, 3790.
- NiknamK,Zolfigol MA,Khorramabadi-ZadA,ZareR,ShayeghM,Catal. Commun. 2006, 7, 494.
- NiknamK,KaramiB,ZolfigolMA,Catal. Commun.2007, 8, 1427.
- NiknamK,SaberiD,NouriSefatM,Tetrahedron Lett.2009, 50, 4058.
- KNiknam, SaberiD,MohagheghnejadM, Molecules, 2009, 14, 1915.
- YoonH,KimJ,LeeN,KimB,JangJ,Chembiochem, 2008, 9, 634.
- AntoliniE,GonzalezER,Appl. Catal. A: Gen. 2009, 365,1.
- XiaoR,ChoSII,LiuR,LeeSB,J. Am. Chem. Soc.2007, 129, 4483.
- MalekAbbaslouRM,SoltamJ,Dalai AK,Appl. Catal. A: Gen. 2010, 379, 129.
- LeeJS, LuoH, Baker G,Dai A S, Chem. Mater.2009, 21, 4756.
- Qiu,L.; Liu,B.;Peng, Y.; Yan,F.,Chem. Commun.2011, 47, 2934.
- Chu, F.; Lin, B.; Yan, F.;Qiu, L.; Lu, J.,J. Power Sources.2011, 196, 7979.
- Kuo S C, Huang L J and Nakamura H.,J. Med. Chem. 1984, 27 539
- Wang J-L, Liu D, Zhang Z-J, Shan S, Han X, Srinivasula S M, Croce C M, Alnemri E S and Huang Z Proc. Natl. Acad. Sci. USA 2000, 97,7124
- Zaki M E A, Soliman H A, Hiekal O A and Rashad A E . Naturforsch.2006, 61,1
- Zhu J and BienaymeH Multicomponent reactions, Weinheim: Wiley-VCH, 2005
- Balaskar R S, Gavade S N, Mane M S, Shingate B B, Shingare M S and Mane D V Chin. Chem. Lett.2010, 21,1175
- Mecadon H, Rohman M R, Kharbangar I, Laloo B M, Kharkongor I, Rajbangshi M and Myrboh B TetrahedronLett. 2011, 52,3228
- Heravi M M, Ghods A, Derikvand F, Bakhtiari K and Bamoharram F FJ. Iran Chem. Soc. 2010, 7,615
- Vasuki G and KumaravelK TetrahedronLett. 2008, 49,5636
- Gogoi S and Zhao C-G TetrahedronLett. 2009, 50,2252
- Sheibani H and BabaieM Synth. Commun. 2010, 40,257
- Mohammadi-Ziarani G, Abbasi A, Badiei A and Aslani Z,E-J. Chem. 2011, 8,293
- Gao S, Tsai C H, Tseng C and Yao C-F,Tetrahedron 2008, 64,9143
- Fotouhi L, Heravi M M, Fatehi A and Bakhtiari K,Tetrahedron Lett. 2007, 48,5379
- Azaifar D, Khatami S-M and Nejat-Yami R,J. Chem. Sci. 2014, 126,95
- Azarifar A, Nejat-Yami R, Al-Kobaisi M and Azarifar D,J. Iran Chem. Soc. 2013, 10,439
- Khaksar S, Rouhollahpour A and Talesh S M,J. Fluorine Chem. 2012, 141,11
- TianX,Sub F, and ZhaoX S,Green Chem.2008, 10, 951.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


