Reaction of 1-Carboxymethyluracil with Amino acid alkyl ester hydrochloride
N. Srivastava1*, A. Mehrotra1
1Bioorganic and Heterocyclic Research Laboratory, Department of Applied Chemistry, Faculty of Engineering and Technology, M.J.P. Rohilkhand University, Bareilly-243006 U.P. India.
DOI : http://dx.doi.org/10.13005/ojc/310172
Article Received on :
Article Accepted on :
Article Published : 28 Feb 2015
We have synthesized derivatives of uracil 3 by the reaction of 1-carboxymethyl uracil 1 and different amino acid alkyl ester hydrochloride 2 in the presence of Et3N, DCC, HOBt. The structure of synthesized compounds was characterized by IR, 1H NMR and mass spectroscopy.
KEYWORDS:1-carboxymethyluracil; amino acid alkyl ester hydrochloride
Download this article as:| Copy the following to cite this article: Srivastava N, Mehrotra A. Reaction of 1-Carboxymethyluracil with Amino acid alkyl ester hydrochloride. Orient J Chem 2015;31(1). |
| Copy the following to cite this URL: Srivastava N, Mehrotra A. Reaction of 1-Carboxymethyluracil with Amino acid alkyl ester hydrochloride. Orient J Chem 2015;31(1). Available from: http://www.orientjchem.org/?p=7493 |
Introduction
Heterocyclic compounds [1, 2] have received considerable attention due to their wide range of pharmacological activities. Pyrimidine is most important heterocyclic compound with two nitrogen atoms. Different Pyrimidine derivatives [3] comprises of interesting medicinal properties. Uracil is among the five nucleobases like adenine, guanine, cytosine, and thymine but is only found in RNA. 5-Fluorouracil [4] is found to be antimetabolite of uracil and is one of most widely used antineoplastic agents. Antimicrobial properties of various nitro uracil derivatives have been studied. Peter G. Schultz et al [5] have reported the preparation of 1-carboxymethyluracil methyl glycinate amide and 1-carboxymethyluracil glycine amide by the photocleavage reaction of uracil dimer.
By considering the above facts we have started our work on the synthesis of uracil derivatives and therefore reporting here a method for the synthesis of uracil derivative 3.
Chemistry
The first step involves the preparation of 1-carboxymethyluracil by the usual reported method [5-7]. The synthesis of the 1-carboxymethyluracil alkyl glycinate amide derivatives 3 was carried out by the reaction of 1-carboxymethyluracil 1 and amino acid alkyl ester hydrochloride 2 at 0°C in the presence of Et3N, DCC/HOBt as the coupling reagent [8-10] .The desired compounds 3 was isolated as the sole product in good yield. Scheme 1 represents the sequence of reaction for the synthesis of the desired compounds 3.The structure of all the synthesized compounds were characterized by means of IR, 1H NMR and mass spectra.
Result and Discussion
The present work describes the synthesis of 1-carboxymethyluracil alkyl glycinate amide derivatives 3 synthesized from 1-carboxymethyluracil 1 and amino acid alkyl ester hydrochloride 2 in the presence of Et3N, DCC/HOBt. The structure of the synthesized compounds 3 was characterized by means of IR, 1H NMR, mass spectra and all data are in accordance with the assigned structure 3.
Experimental
Spectral data were recorded as follows: IR Spectra was run on a Perkin Elmer and Schimadzu 8201 PC, FT Infrared spectrophotometer (ν in cm-1). Mass spectra were recorded on Jeol SX-102 (FAB). 1H NMR has been recorded on Bruker Avance-300 (300 MHz) and chemical shifts (δ in ppm) were reported relative to the solvent peak (CDCl3+DMSO-d6) or TMS as internal standard. Signals were designated as follows: s, singlet; bs, broad signal; d, doublet; t, triplet; m, multiplet. Melting points were determined by open capillary method and were uncorrected. All reagents used were of commercial grade and used as received without further purification unless otherwise specified. Dry DMF was used wherever required. Reagent grade solvents were used in all other cases unless otherwise specified. Organic solutions were dried over anhydrous Na2SO4 and concentrated at reduced pressure.
General Procedure for Synthesis of 1-Carboxymethyluracil Alkyl Glycinate Amide 3 A-B
To a well stirred solution of amino acid alkyl ester hydrochloride 2 (12 mmol) suspended in anhydrous DMF (12 mL) neutralized with triethylamine in portions (12 mmol) at 0oC, 1-caboxy methyluracil 1 (12 mmol) and 1-hydroxy benzotriazole (12 mmol), dissolved in dry DMF (8 mL) were added consequently in the reaction flask in the small portions. The mixture was stirred at the same temperature for few min and dicyclohexyl carbodiimide (DCC, 12 mmol) dissolved in dry DMF (16 mL) was added drop wise in 30 min under the same condition. Stirring was continued in ice-cold condition for 4 h and left for overnight at the room temperature. The precipitated dicyclohexylurea (DCU) was filtered off and washed with DMF and combined filtrates were evaporated to dryness under reduced pressure. The solid residue was triturated with ethyl acetate and the resulting organic layer was washed with H2O (2×60 mL), ice-cold 1N HCl (3×60 mL), water (2×60 mL), saturated NaHCO3 (3×60 mL) and finally with H2O (3×80 mL). The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to get the desired product.
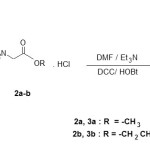 |
Scheme 1 Click here to View scheme |
Spectral characterization of synthesized compounds 3a and 3b are given below
Carboxymethyluracil Methyl Glycinate Amide (3a)
Yield: 90%; mp: 190 0C; elemental analysis: Anal. Calcd. for C9H11N3O5: C,44.82, H,4.60, and N,17.42, Found: C,44.70, H,4.81, and N,17.11; IR (KBr, cm-1 ): 301, 3015, 2825, 1740, 1680, 1550, 1450, 1225, 1080, 740, 625 cm-1; 1H-NMR (300 MHz,CDCl3+DMSO-d6) δ(ppm): 3.52 (s, 3H, -OCH3), 3.84(d, 2H, -CH2-), 4.40 (s, 2H, -CH2-), 5.50 (d, 1H, Ar-H) , 7.50 (d, 1H, Ar-H),8.60 (bs, ,1H ,NH) ,11.10 (s, 1H, NH); MS m/z 241 (M+).
Carboxymethyluracil Ethyl Glycinate Amide (3b)
Yield: 85%; mp: 210 0C; elemental analysis: Anal. Calcd. for C10H13N3O5:C,47.06, H,5.13, N,16.46; found: C,47.45, H,5.51, N,16.23; IR (KBr, cm-1):3425, 3300, 3125, 2805, 1700, 1665, 1566, 1430, 1228, 1005, 752, 650; 1H-NMR (300 MHz, CDCl3+DMSO-d6) δ(ppm): 1.20(t,3H, -CH2CH3), 3.70(d,2H,-CH2),4.15 (q, 2H, -OCH2CH3), 4.40 (s, 2H, -CH2-), 5.40 (d, 1H, Ar-H) ,7.30 (d, 1H, Ar-H), 8.33 (bs, 1H, NH), 10.50(s, 1H, NH); MS m/z 255 (M+).
Acknowledgements
This work was financially supported by the University Grant Commission, New Delhi (Grant no 31-128/2005 SR). We thank to Sophisticated Analytical Instrument Facility (SAIF), Central Drug Research Institute, Lucknow-226001(India) for providing spectral data on payment basis.
References
- Fascio, M.L.; Errea, M. I. ; Accorso, N. B. D’. Eur. J. Med.Chem. 2015, 90, 666-683.
- Plech, T.; Wujec, M.; Kosikowska U.; Malm A.; Rajtar B.; Polz-Dacewicz M. Eur. J. Med. Chem. 2013, 60, 128-134.
- Bekhit, A.A. ; Fahmy H. T.Y. ; Rostom S. A. F. ; Baraka A. M. Eur. J. Med. Chem. 2003, 38, 27-36.
- Kingsbury W.D.; Boehm J. C. ; Mehta R. J. ; Grappel S.F.; Gilvarg C. J. Med. Chem. 1984, 27 , pp 1447–1451.
- Jacobsen, J. R.; Cochran, A. G.; Stephans, J. C.; King, D. S.; Schultz, P. G. J. Am. Chem. Soc. 1995, 117, 5453-5461.
- Wheeler, H. L.; Liddle, L. M. J. Am. Chem. Soc. 1908, 30, 1152-1156.
- X. J. Liu, G. C. Chi, and R. Y. Chen, Chin. J. Synth. Chem. 1999, 7, 219.
- Ibrahim, A.I. A.; Imran, A. Al-M.; Bahjat. S.; Najim, A. Al-M.; Palo, L.C. Heteroatom Chem. 2005, 16, 148.
- Sanda, F.; Nakamura, M.; Endo, T. Macrommolecules 1996, 29, 8064.
- Katritzky, A.R.; Narindoshvili, T. Org Biomol Chem. 2008, 6, 3171.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


