Solvent-free synthesis of stable oxaphosphaphenanthrene derivatives using multicomponent reaction of naphthols
Sara Hallajian1, Eskandar Alipour1, Naser Foroughifar1 and Mohammad A. Khalilzadeh2
1Department of Chemistry, Islamic Azad University, Tehran North Branch, Tehran, Iran
2Department of Chemistry, Qaemshahr Branch, Islamic Azad University, Qaemshahr, Iran
DOI : http://dx.doi.org/10.13005/ojc/300348
Article Received on :
Article Accepted on :
Article Published : 23 Aug 2014
The reaction of dimethyl acetylenedicarboxylate with OH-acid such as 2-naphthol in the presence of trimethyl or triphenyl phosphite under solvent-free conditions produce oxaphosphaphenanthrene derivatives in good yields. Also, the reaction of dimethyl acetylenedicarboxylate and trimethyl phosphite in the presence of 1-naphthol leads to succinate derivatives in excellent yields. Using triethyl phosphite and dimethyl acetylenedicarboxylate in the presence of 1-naphthol or 1-naphthol produces chromene-4-carboxylate derivatives in good yields.
KEYWORDS:Dimethyl acetylenedicarboxylate; Solvent-free conditions; oxaphosphaphenanthrene; 1-naphthol
Download this article as:| Copy the following to cite this article: Hallajian S, Alipour E, Foroughifar N, Khalilzadeh M. A. Solvent-free synthesis of stable oxaphosphaphenanthrene derivatives using multicomponent reaction of naphthols. Orient J Chem 2014;30(3). |
| Copy the following to cite this URL: Hallajian S, Alipour E, Foroughifar N, Khalilzadeh M. A. Solvent-free synthesis of stable oxaphosphaphenanthrene derivatives using multicomponent reaction of naphthols. Orient J Chem 2014;30(3). Available from: http://www.orientjchem.org/?p=4319 |
Introduction
Multi-component reactions (MCRs), due to their productivity, simple procedures, convergence, and facile execution, are one of the best tools in combinatorial chemistry [1]. Therefore, the design of novel MCRs has attracted great attention from research groups working in areas such as drug discovery, organic synthesis and materials science. As a result, the number of new MCRs has grown rapidly [2]. Green chemistry approaches hold out significant potential not only for reduction of byproducts, a reduction in the waste produced, and lowering of energy costs but also in the development of new methodologies toward previously unobtainable materials, using existing technologies [3-5]. Organophosphorus compounds, i.e. those bearing a carbon atom directly bound to a phosphorus atom, are synthetic targets of interest, not least because of their value for a variety of industrial, biological, and chemical synthetic uses [6-8]. As a result, a large number of methods have appeared describing novel syntheses of organophosphorus compounds. There are many studies on the reaction between trivalent phosphorus nucleophiles and α,β-unsaturated carbonyl compounds in the presence of a proton source such as alcohol or phenol [9-11]. We describe herein the reaction of dimethyl acetylenedicarboxylate with triphenyl, triethyl and trimethylphosphit as the P-nucleophile in the presence of OH-acid such as 1-naphthol or 2-naphthol under solvent-free conditions at 70 oC in excellent yield (Scheme 1).
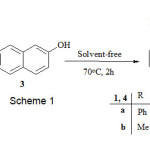 |
Scheme 1 Click here to View Scheme |
Material and methods
Melting points were measured on an Electrothermal 9100 aparatus. Elemental analyses for the C, H, and N were performed using a Heraeus CHN-O-Rapid analyzer. Mass spectra were recorded on a FINNIGAN-MATT 8430 spectrometer operating at an ionization potential of 70 eV. IR spectra were measured on a Shimadzu IR-460 spectrometer. ¹H , ¹³C, and ³¹P NMR spectra were measured with a BRUKER DRX-500 AVANCE spectrometer at 500.1, 125.8, and 202.4 MHz, respectively. 1H, 13C, and 31P spectra were obtained for solutions in CDCl3 using TMS as internal standard or 85% H3PO4 as external standard. All the chemicals used in this work were purchased from Fluka (Buchs, Switzerland) and are used without further purification.
General procedure for preparation of compounds 4a-b and 9, 17.
Dimethyl2,2-diphenoxy-4H-1-oxa-2l5-phosphaphenanthrene-3,4-dicarboxylate (4a).
To a magnetically stirred of 0.28 g dimethyl acethylenedicarboxylate 2 (2 mmol) and 0.29 g 2-naphthol 3 (2 mmol) was added 0.62 g triphenyl phosphite 1a (2 mmol) at 70oC. The reaction mixture was then stirred for 2 h. The reaction mixture was crystallized from diethyl ether. The product 4a was obtained as colorless crystals, m.p. 178-180 °C, 0.90 g, yield 90%. IR (KBr) (νmax/cm-1): 1667, and 1722 (2 C=O). Anal. Calcd for C28H23O7P (502.4): C, 66.93; H, 4.61. Found: C, 66.9; H, 4.7%. 1H NMR (500 MHz, CDCl3): δ 3.21 and 3.72 (6 H, 2 s, 2 Me), 5.55 (1 H, d 3JHP 33 Hz, CH), 6.98-9.20 (16 H, m, 16 CH). 13C NMR (125.7 MHz, CDCl3): δ 41.10 (d, 1JCP 225 Hz, C), 41.37 (d, 2JCP 9 Hz, CH), 50.32 and 52.06 (2 OMe), 118.22 (d, 3JPC 6 Hz, CH of C10H6), 120.54 (d, 3JCP 4 Hz, 2 CHortho of Ph), 120.83 (d, 3JPC 9 Hz, C of C10H6), 121.50 (d, 3JPC 4 Hz, 2 CHortho of Ph), 124.10, and 125.56 (2 CH of C10H6), 125.92 and 126.14 (2 CHpara of Ph), 127.49 and 128.43 (2 CH of C10H6), 129.71 (m, 4 CHmeta of Ph groups), 129.79 (CH of C10H6), 131.13 and 131.33 (2 C of C10H6), 148.13 (d 2JPC 8 Hz, C-O of C10H6), 149.96 (m, 2 Cipso of Ph groups), 168.40 (d 2JPC 17.3 Hz, C=O), 173.32 (C=O). 31P NMR (202 MHz, CDCl3): δ 41.54.
Dimethyl 2,2-dimethoxy-4H-1-oxa-2l5-phospha-phenanthrene-3,4-dicarboxylate (4b).
The procedure for preparation of 4b was similar to that for 4a. Colorless crystals, m.p. 128-130 °C, 0.64 g, yield 85%. IR (KBr) (νmax/cm-1): 1652, and 1722 (2 C=O). Anal. Calcd for C18H19O7P (378.3): C, 57.15; H, 5.06. Found: C, 57.2; H, 5.1%. 1H NMR (500 MHz, CDCl3): δ 3.61 and 3.70 (6 H, 2 s, 2 Me), 3.65 (3 H, d 3JPH 13 Hz, OMe), 4.07 (3 H, d 3JPH 13 Hz, OMe), 5.65 (1 H, d 3JHP 31 Hz, CH), 7.26 (d, 3JHH 9 Hz, CH), 7.45 (1 H, t, 3JHH 9 Hz, CH), 7.59 (1 H, t, 3JHH 9 Hz), 7.73 (1 H, d 3JHH 9 Hz, CH), 7.79 (1 H, d 3JHH 9 Hz, CH), 8.39 (1 H, d 3JHH 9 Hz, CH). 13C NMR (125.7 MHz, CDCl3): δ 39.20 (d, 1JCP 222 Hz, C), 41.37 (d, 2JCP 9 Hz, CH), 50.37 and 52.21 (2 OMe), 55.33 (d, 2JPC 6 Hz, P-OMe), 55.65 (d, 2JPC 5 Hz, P-OMe), 118.35 (d, 3JPC 6 Hz, CH), 121.29 (d, 3JPC 9.1 Hz, C), 124.40, 125.43, 127.38, 128.40 and 129.79 (5 CH), 131.13 and 131.31 (2 C), 148.58 (d 2JPC 8 Hz, C-O), 169.20 (d 2JPC 18 Hz, C=O), 174.92 (C=O). 31P NMR (202 MHz, CDCl3): δ 42.53.
Methyl 3-oxo-2,3-dihydro-1H-benzo[f]chromene-1-carboxylate (9).
To a magnetically stirred of 0.28 g dimethyl acethylenedicarboxylate 2 (2 mmol) and 0.29 g 2-naphthol 3 (2 mmol) was added 0.33 g triethyl phosphite 1c (2 mmol) at 70 oC. The reaction mixture was then stirred for 5 h. The reaction mixture was crystallized from diethyl ether. (0.49 g, yield 96%). m.p. 151-152 °C, IR (KBr) (νmax/cm-1): 1711, and 1752 (2 C=O). Anal. Calcd for C15H12O4 (256.3): C, 70.31; H, 4.72. Found: C, 70.1; H, 4.7%. 1H NMR (500 MHz, CDCl3): δ 2.82 (1 H, dd 2JHH 16 Hz 3JHH 6 Hz, HCH), 3.25 (1 H, d 2JHH 16 Hz, HCH), 3.65 (3 H, s, OMe), 4.60 (1 H, d 3JHH 6 Hz, CH), 7.26 (d, 3JHH 9 Hz, CH), 7.49 (1 H, t, 3JHH 8 Hz, CH), 7.59 (1 H, t, 3JHH 8 Hz), 7.73 (1 H, d 3JHH 8 Hz, CH), 7.79 (1 H, d 3JHH 8 Hz, CH), 8.32 (1 H, 3JHH 9 Hz, CH). 13C NMR (125.7 MHz, CDCl3): δ 31.37 (CH2), 37.87 (CH), 52.84 (OMe), 112.73 (C), 117.70, 123.27, 125.42, 127.75, 128.76, and 130.65 (6 CH), 130.90, 131.00, and 149.96 (3 C), 166.16 and 171.22 (2 C=O). MS, m/z (%): 256 (M+., 15), 196 (100), 168 (41).
Dimethyl2-(dimethoxy-phosphoryl)-3-(1-hydroxy-naphthalene-2-yl)-succinate (17).
To a magnetically stirred of 0.28 g dimethyl acethylenedicarboxylate 2 (2 mmol) and 0.29 g 1-naphthol 16 (2 mmol) was added 0.25 g trimethyl phosphite 1b (2 mmol) at 70 oC. The reaction mixture was then stirred for 4 h. The reaction mixture was crystallized from diethyl ether. The product 17 was obtained as colorless crystals, m.p. 173-185 °C, 0.76 g, yield 96%. IR (KBr) (νmax/cm-1): 3230 (OH), 1724 (C=O). Anal. Calcd for C18H21O8P (396.3): C, 54.55; H, 5.34. Found: C, 54.4; H, 5.3%. 1H NMR (500 MHz, CDCl3): δ 2.85 (3 H, d 3JHP 11 Hz, OMe), 3.60 (3 H, s, OMe), 3.67 (3 H, d 3JHP 11 Hz, OMe), 3.82 (3 H, s, OMe), 3.88 (1 H, dd 2JHP 21 Hz 3JHH 12 Hz, CH), 4.99 (1 H, dd 3JHH 12 Hz 3JHP 9 Hz, CH), 7.10-8.42 (6 H, m, 6 CH), 8.47 (1 H, s, OH). 13C NMR (125.7 MHz, CDCl3): δ 44.04 (CH), 48.51 (d 1JPC 135 Hz, CH), 52.79 (OMe), 52.86 (d 2JPC 7 Hz, OMe), 53.05 (OMe), 53.97 (d 2JPC 7 Hz, OMe), 116.92 (C), 121.78 and 123.42 (2 CH), 124.60 (C), 125.74, 126.82 and 127.20 (3 CH), 127.67 (C), 134.20 (CH), 150.50 (C), 168.24 (d 2JPC 5 Hz, C=O), 173.01 (d 3JPC 22 Hz, C=O). 31P NMR (202 MHz, CDCl3): δ 18.56. MS, m/z (%): 396 (M+., 42), 305 (82), 273 (100), 168 (87),139(72).
Result and discussion
The reaction of dimethyl acetylene dicarboxylate 2 and 2-naphthol 3 in the presence of trimethyl or triphenyl phosphite 1 leads to oxaphosphaphenanthrene-3,4-dicarboxylate derivatives 4 in good yields (Scheme 2). The structures of 4 were determined on the basis of their 1H, 13C, and 31P NMR spectra, IR spectra, elemental analyses, and mass spectrometric data. The 1H NMR spectrum of 4a in CDCl3 shows two singlets at δ = 3.21 and 3.72 ppm for the methoxy protons and one doublet at δ = 5.48 (3JPH = 34 Hz) for the methine proton, along with multiplets at δ = 6.98-9.20 for the aromatic protons. Characteristic carbonyl resonances appear clearly at δ = 168.40 (d, 2JPC = 17 Hz), 173.32 ppm, whereas the ylide carbon atom exhibits resonances at δ = 41.10 (d, 1JPC = 225 Hz) ppm. The observed 1JCP values are typical of an α-ylide ester [12]. The double bond character of the C-P bond and the presence of three electronegative oxo substituent on the phosphorus atom increases the 1JCP value.11 Evidence for the presence of an oxaphosphaphenantrene skeleton in 4a was shown by the 13C signals at δ = 118.22 (d, 3JCP = 6 Hz) for CH of naphthalene moiety and at δ = 148.13 (d, 2JCP = 8 Hz) for the C-O carbon of naphthalene. 31P NMR signals was found at δ = 41.54 ppm. The 1H and 13C NMR spectra of 4b were similar to those for 4a except for the phosphoranyl moiety. Although we have not yet established the mechanism of the reaction between dimethylacetylene dicarboxylate and phosphites in the presence of 2-naphthol in an experimental manner, a possible explanation is proposed in (Scheme 2). On the basis of the well established chemistry of phosphorus nucleophiles2,3 it is reasonable to assume that ylide 4 results from initial addition of the phosphite to DMAD and subsequent protonation of the reactive 1:1 adduct, followed by attack of carbon atom of the anion of 2-naphthol 6 to cation 5 to generate ylide 7 which isomerises under the reaction conditions employed to produce the oxaphosphorane 8. Elimination of ROH from 8 leads to product 4. The reaction between triethyl phosphite 1, dimethylacetylene dicarboxylate 2, and 2-naphthol 3 quantitatively gave product 9 (Scheme3).
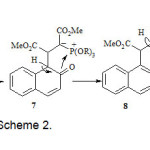 |
Scheme 2 Click here to View Scheme |
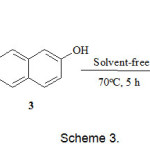 |
Scheme 3 Click here to View Scheme |
The IR spectrum of 9 exhibited the ester and lactone carbonyl groups at 1711 and 1752 cm-1, respectively. The 1H NMR spectrum of 9 showed a double doublet at δ = 2.82 (2JHH = 16 Hz, 3JHH = 6 Hz) for one of the methylene protons. The other methylene proton displayed a doublet (2JHH = 16 Hz) at δ = 3.25 ppm. The methine proton showed a doublet (3JHH = 6 Hz) at δ = 4.60 ppm. The coupling constants observed for this AMX system are consistent with a conformation in which theH-C-C-H dihedral angles for the CH-CH2 moiety are expected to be about 90º and 30º [12]. The 13C NMR spectrum of 9 displayed 15 distinct resonance in agreement with the proposed structure. A possible mechanism for the formation of compound 9 is shown in Scheme 4. The oxaphosphorane 12 is formed in similar steps shown for oxaphosphorane 8 in Scheme 2. However, since the ethoxide anion is a weaker living group, cleavage of the phosphorus-oxygen bond of the naphthol residue become favorable, giving dimethyl succinate 15. Lactonization of this hydroxy-ester gave product 9.
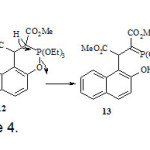 |
Scheme 4 Click here to View Scheme |
Under similar conditions, the reaction of dimethylacetylene dicarboxylate 2 and trimethyl phosphite 1 in the presence of 1-naphthol 16 gave succinate 17 in good yield (Scheme 5).
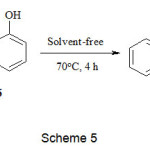 |
Scheme 5 Click here to View Scheme |
The 1H NMR spectrum of 17 displayed signals for vicinal methine protons at δ = 3.88 and 4.99, which appeared as two set of double doublets with 2JHP and 3JHP values of 21 and 9 Hz, respectively. The methoxy groups of the phosphoranyl moiety are diastereotopic and show two separate doublets at δ = 2.85 and 3.67. The hydroxy proton was observed as a broad singlet at δ = 8.47 which disappeared with addition of D2O. Observation of 3JHH = 12 Hz for the vicinal methine protons in 17 indicates the dominance of anti arrangement. A proposed mechanism for the formation of compound 17 is shown in Scheme 4.
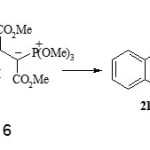 |
Scheme 6 Click here to View Scheme |
In conclusion, we found that the reaction of dimethylacetylene dicarboxylate with trimethyl phosphite or triphenyl phosphite in the presence of 2-naphthol leads to functionalizedoxaphosphaphenanthrenes. The reaction of 1-naphthol with dimethylacetylene dicarboxylate and trimethyl phosphite produces, stereoselectively benzochromene derivative in high yield. The addition reaction of triethyl phosphite, dimethylacetylene dicarboxylate, and 1-naphthol or 2-naphthol leads quantitatively to benzochromene derivatives. The present method carries the advantage that, not only is the reaction performed under solvent-free conditions, but also the substances can be mixed without any activation or modification.
References
- Terrett N. K., Combinatorial Chemistry, Oxford University Press. New York,(1998)
- (a) Orru, R. V. A.; Greef, M.Synthesis. 2003,10 ,1471–1499, (b) Do¨mling, A.Curr. Opin. Chem. Biol.2002, 6, 306–313, (c) Bienayme, H.; Hulme, C.; Oddon, G.; Schmitt, P.Chem. Eur. J. 2000,6, 3321–3329, (d) Do¨mling, A. Chem. Rev.2006, 106, 17–89, (e) Zhu J. and Bienayme´ H.,Multicomponent Reactions, Wiley-VCH.weinheim, (2005)
- Anastas P. andWilliamson T., Green Chemistry: Frontiers in Benign ChemicalSynthesis and Procedures, Oxford Science Publications. New York, (1998)
- Cave, G. W. V.; Raston, C. L.; Scott, J. L.Chem. Commun. 2001,21, 2159-2169
- Sheldon R. A., As estimated by determination of E-factor,Vol. 12.; Chem. Ind. (1997)
- Corbridge D. E. C., Phosphorus: An Outline of Its Chemistry, Biochemistry and Uses, 5th ed.;Elsevier. Amesterdam, (1995)
- Engle R., Synthesis of Carbon-Phosphorus Bond, CRC Press. Boca Raton, FL, (1988)
- Cadogan J. I. G., Organophosphorus Reagents in Organic Synthesis, Academic Press. New York, (1977)
- Hudson H. R., in The Chemistry of Organophosphorus Compounds,Primary,Secondary and Tertiary Phosphines, Polyphosphines and Heterocyclic Organophosphorus(III) Compounds, Vol. I.; John Wiley. New York, (1990)
- George, M. V.; Khetan, S. K.; Gupta, R. K.Adv. Heterocycl. Chem.1976, 19, 354-355
- Burgada, R.; Leroux, Y.; Zabloka, M.; El Khoshnieh, Y. U. Tetrahedron Lett.1981,22, 33-35
- Silverstein R. M., Bassler, C. G. and Morrill T. C., Spectroscopic Identification of Organic Compounds, 5th ed.;John Wiley. New York, (1992)

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


