Synthesis of novel xylofuranosyloxymethyl nucleosides
1Organic Division III, Indian Institute of Chemical Technology, Tarnaka, Hyderabad, AP, India 500 007
2Current address: Institute for Molecular Bioscience, University of Queensland, St Lucia, QLD, Australia 4072
3Current address: Sri Krishna Pharmaceuticals, C-4 Industrial Area, Uppal, Hyderabad, AP, India 500 026
DOI : http://dx.doi.org/10.13005/ojc/300202
Download this article as:
![]()
The ability to selectively trap xylofuranosyloxy carbocation instead of the xylofuranosyl cation by activation of acetoxymethoxy leaving group at the anomeric centre is demonstrated for the synthesis of xylofuranosyloxymethyl nucleosides. The use of SnCl4 as an activator gives both xylofuranosyloxymethyl and natural nucleosides, whereas use of TMSOTf exhibits selectivity to give xylofuranosyloxymethyl nucleosides in case of uracil, thymine, guanine and a mixture of nucleosides in case of cytosine and adenine.
KEYWORDS:Glycosyloxymethyl nucleosides; Lewis acid; Glycosylation; Thymine; Adenine.
INTRODUCTION
The chemistry of nucleosides1 was revolutionized after the discovery of modified nucleosides2 as effective therapeutic agents for the treatment of AIDS and cancer.3,4 Examples of modified nucleosides approved by US Food and Drug administration are: 3’-azidothymidine (AZT), dideoxycytidine (ddC), dideoxyinosine (ddI), 2’,3’-didehydro-2’,3’-dideoxythymidine (d4T), 3’-azido-2’,3’-dideoxyuridine (AzddU).5 Another important class of sugar modified nucleosides are acyclic nucleosides, where a sugar unit is replaced by acyclic group.6 The first acyclic nucleoside 9-[(2-hydroxyethoxy) methyl] guanine (ACV) is a highly specific inhibitor of herpes simplex virus proliferation (Figure 1). Ganciclovir (GCV), structurally different from acyclovir by the addition of a hydroxymethyl group to the acyclic side chain is effective for cytomegalovirus infections.
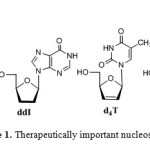 |
Figure 1: Therapeutically important nucleoside analogues |
Glycosylation reaction 7 is crucial for the synthesis of complex glycoconjugates, including the nucleosides. Although there has been fairly significant advancement in the field of glycoconjugate synthesis, the efficiency of glycosidic bond formation in achieving high chemical yield, stereoselectivity and regioselectivity8 still remains challenging.9 The classical aspects of carbohydrate chemistry centers around the anomeric carbon where glycosidic bond formation takes place via glycosyl cation10 and stereochemical outcome depends on attaching the aglycones to give either α– or β–glycosides.11 We were interested in developing efficient glycosylation methods for the synthesis of glycoconjugates.12 In continuation of our efforts to develop efficient glycosylation reactions,we designed an acetoxymethoxy leaving group at the anomeric centre and demonstrated its ability as glycosyl donor.13 During our investigations, we found that when a glycosylation reaction was performed with O-nucleophiles, it resulted into O-glycosides, whereas N-nucleophiles afforded glycosyloxymethyl derivatives.13,14 The formation of glycosyloxymethyl derivatives was explained based on capturing the glycosyloxymethyl cation ‘b’ by N-nucleophiles such as nucleic acid bases (Scheme 1).14 In order to confer generality to the concept developed for synthesis of novel nucleosides and to probe the reactivity of other glycosyl donors we report the activation of xylofuranosyl donor. Furthermore, given the medicinal importance of acylic nucleosides, synthesis of modified glycosyloxymethyl nucleosides may provide new therapeutic agents that can inhibit herpes simplex virus.
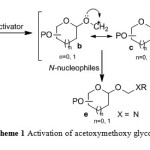 |
Scheme 1: Activation of acetoxymethoxy glycosyl donor |
RESULTS AND DISCUSSION
The key substrate xylofuranosyl donor 4 was prepared starting from 1-O-acetyl-2,3,5-tri-O-benzoyl-α/β-D-xylofuranoside (1).14 Compound 1 on reaction with propargyl alcohol and BF3.OEt2 at room temperature gave 2-propynyl 2,3,5-tri-O-benzoyl-β-D-xylofuranoside (2) (Scheme 2).15 Compound 2 on further reaction with a catalytic amount of Hg(OCOCF3)2 in acetone-H2O (2:1) at room temperature gave the corresponding 2-(oxopropoxy)-2,3,5-tri-O-benzoyl-β-D-xylofuranoside (3). Baeyer-Villiger rearrangement of 3 with m-CPBA in CHCl3 at room temperature gave 1-[(2,3,5-tri-O-benzoyl-β-D-(xylofuranosyloxy)methyl]acetate (4) in 45% overall yield from compound 1.Compound 4 was characterized by 1H NMR spectrum from the appearance of methylene protons at δ 5.40-5.48 (m, 2H), anomeric proton (H-1) at δ 5.50 as a singlet indicating the β-anomeric configuration and by 13C NMR from the appearance of methylene carbon at δ 85.1 and anomeric carbon (C-1) at δ 104.9.
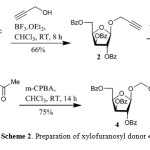 |
Scheme 2: Preparation of xylofuranosyl donor 4 |
To envisage the concept, we first investigated the coupling reactions of xylofuranosyl donor 4 with pyrimidine bases 5-8.16 Thus, coupling reaction of 4 with 5 in CHCl3 and using activator SnCl4 at room temperature gave 1-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]uracil (9) and 1-(2’,3’,5’-tri-O-benzoyl-β-D-xylofuranosyl)uracil (10) in a ratio of 1.0:1.9 (78% yield) (Scheme 3). A similar coupling reaction of 4 with bis(trimethylsilyl)thymine (6) by use of SnCl4 gave 1-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]thymine (12) and 1-(2’,3’,5’-tri-O-benzoyl-β-D-xylofuranosyl)thymine (13) in a ratio of 1.0:2.1 (66% yield) (Scheme 3). Coupling reaction of 4 with bis(trimethylsilyl)cytosine (7) by use of SnCl4 gave 1-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]cytosine (15) and 1-(2’,3’,5’-tri-O-benzoyl-β-D-xylofuranosyl)cytosine (16) in a ratio 1.0:2.4 (62% yield) respectively (Scheme 3).
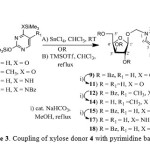 |
Scheme 3: Coupling of xylose donor 4 with pyrimidine bases 5-8 |
The xylofuranosyloxy methyl pyrimidine nucleosides 9, 12, 15 were separated from pyrimidine nucleo sides 10, 13, 16 by column chromatography and characterized by 1H and 13C NMR. The methylene protons, characteristic of xylofuranosyloxymethyl nucleosides 9, 12, 15 appeared at δ 5.20-5.40 (2H, AB type quartet, Jgem = 9.6-11.0 Hz). Compounds 10, 13, 16 were characterized as natural nucleoside by comparison of 1H NMR, melting point and specific rotation with that reported in the literature.16-18 Compound 9, 12, 15 on debenzoylation gave 1-[(β-D-xylofuranosyloxy)methyl]uracil (11), 1-[(β-D-xylofuranosyloxy)methyl]thymine (14), 1-[(β-D-xylofuranosyloxy)methyl]cytosine (17) respectively in good yields. Regiochemistry (N1 or N3 linked nucleosides) of the newly formed xylofuranosyloxymethyl nucleosides 9, 12, 15 were assigned as N-1 linked nucleosides based on the (2 nm or less) absorption difference observed in UV spectra of the compounds 11, 14, 17 in neutral vs. 0.1 N alkali.18
Once we accomplished the synthesis of xylofuranosyloxymethyl pyrimidines, we studied the coupling reactions of 4 with purine bases. Thus, coupling reaction of 4 with bis(trimethylsilyl)-N6-benzoyladenine19 (20) using SnCl4 in MeCN at room temperature resulted in the isolation of N6-benzoyl-9-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]adenine (21) and 9-(2’,3’,5’-tetra-O-benzoyl-β-D-xylofuranosyl)adenine (22) in a ratio of 1.0:2.27 (59%) (Scheme 4). Compound 21 was characterized as xylofuranosyloxymethyl adenine by 1H NMR and nucleoside 22 was characterized as N9-isomer by comparison of its 1H NMR, melting point and specific rotation with that of reported in the literature.18b Compound 21 on debenzoylation gave 9-[(-β-D-xylofuranosyloxy)methyl]adenine (23) and the regiochemistry was assigned based on 1H NMR spectrum from the appearance of H-2 at δ 8.30 and H-8 at δ 8.28 (Scheme 4). A chemical shift difference of ∆δ 0.02 ppm between H-2 and H-8 observed in the 1H NMR spectrum is characteristic of N9 regioisomer.20
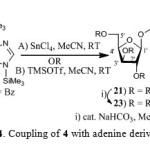 |
Scheme 4: Coupling of 4 with adenine derivative 20 |
Coupling of 4 with guanine derivative 24 using SnCl4 in MeCN at room temperature resulted in the isolation of oxymethylguanine nucleosides, N2-acetyl-9-[(2,3,5-tri-O-benzoyl-β-D-xylofuran- -osyloxy)methyl]guanine (25), N2-acetyl-7-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)methyl] -guanine (26) and natural nucleosides, N2-acetyl-9-(2’,3’,5’-tri-O-benzoyl-β-D-xylofuranosyl) guanine (27) and N2-acetyl-7-(2’,3’,5’-tri-O-benzoyl-β-D-xylofuranosyl)guanine (28). The ratio of xylofuranosyloxymethyl guanine derivatives 25, 26 to natural nucleosides 27, 28 was found to be 1.0:2.2 in 57% yield (Scheme 5). Compound 25, 26 were characterized as xylofuranosyloxymethyl guanine by 1H NMR spectra and compounds 27, 28 were characterized as natural nucleosides by comparison of their 1H NMR spectra with that reported in the literature. Compounds 25, 26 on debenzoylation gave 9-[(-β-D-xylofuranosyloxy)methyl]guanine (29) and 7-[(-β-D-xylofuranosyloxy)methyl]guanine (30) respectively. The regiochemistry of compounds 25, 26 and 29, 30 were assigned by 1H NMR. In case of the compound 29 derived from compound 25, the appearance of H-8 at δ 7.78 as a singlet (1H NMR) and C-5 at δ 116.4 (13C NMR) was characteristic of N9-regioisomer.21 In case of compound 30 derived from compound 26, the appearance of H-8 at δ 8.30 as a singlet (1H NMR) and C-5 at δ 106.5 (13C NMR) was characteristic of N7-regioisomer.21
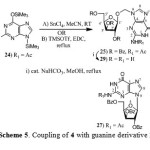 |
Scheme 5: Coupling of 4 with guanine derivative 24 |
In order to probe the reactivity of xylofuranosyl donor 4, we carefully studied the coupling reactions using organic Lewis acid, TMSOTf. Thus, coupling of 4 with 5, 6 by use of TMSOTf afforded the xylofuranosyloxymethyl pyrimidine nucleosides 9 and 12 exclusively. Formation of the corresponding natural nucleosides 10 and 13 were not observed. Coupling of 4 with 7 under similar conditions using TMSOTf showed poor selectivity. It resulted into the formation of xylofuranosyloxymethyl cytosine derivative 15 and natural nucleoside 16 in a ratio of 1.0:1.8. The formation of mixture of cytosine nucleosides may be due to the basicity of cytosine. Therefore, we protected cytosine as N4-bz-cytosine 8 and used for coupling with 4, which resulted in the formation of N4-benzoyl-1-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)-methyl]cytosine (18) in a higher yield compared to natural nucleoside 1-(N4,2’,3’,5’-tetra-O-benzoyl-β-D-xylofuranosyl)cytosine (19) in a ratio of 2.9:1.0. Similarly, coupling of 4 with 20 using TMSOTf as an activator gave xylofuranosyloxymethyl adenine 21 and natural nucleoside 22 in a ratio of 1.0:1.27. Coupling of 4 with 24 using TMSOTf gave oxymethyl guanine nucleosides 25, 26 and natural nucleoside 27, formation of 28 was not observed (Scheme 5). The ratio of natural nucleosides to oxymethyl guanine nucleosides was 1.0:14.0. Thus, synthesis of xylofuranosyloxymethyl nucleosides was achieved by coupling of xylofuranosyl donor 4 with nucleic acid bases similar to ribofuranosyl donor.[14] It was observed that the formation of glycosyloxymethyl nucleosides was in a higher ratio to natural nucleosides in case of ribofuranosyl donor when compared to xylofuranosyl donor during coupling reactions performed using SnCl4. This may be due to subtle difference in the reactivity of xylose donor to the ribose donor. There is no significant difference observed between ribose and xylose donors when activated by TMSOTf.
In order to probe the reactivity of xylofuranosyl donor 4, we carefully studied the coupling reactions using organic Lewis acid, TMSOTf. Thus, coupling of 4 with 5, 6 by use of TMSOTf afforded the xylofuranosyloxymethyl pyrimidine nucleosides 9 and 12 exclusively. Formation of the corresponding natural nucleosides 10 and 13 were not observed. Coupling of 4 with 7 under similar conditions using TMSOTf showed poor selectivity. It resulted into the formation of xylofuranosyloxymethyl cytosine derivative 15 and natural nucleoside 16 in a ratio of 1.0:1.8. The formation of mixture of cytosine nucleosides may be due to the basicity of cytosine. Therefore, we protected cytosine as N4-bz-cytosine 8 and used for coupling with 4, which resulted in the formation of N4-benzoyl-1-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)-methyl]cytosine (18) in a higher yield compared to natural nucleoside 1-(N4,2’,3’,5’-tetra-O-benzoyl-β-D-xylofuranosyl)cytosine (19) in a ratio of 2.9:1.0. Similarly, coupling of 4 with 20 using TMSOTf as an activator gave xylofuranosyloxymethyl adenine 21 and natural nucleoside 22 in a ratio of 1.0:1.27. Coupling of 4 with 24 using TMSOTf gave oxymethyl guanine nucleosides 25, 26 and natural nucleoside 27, formation of 28 was not observed (Scheme 5). The ratio of natural nucleosides to oxymethyl guanine nucleosides was 1.0:14.0. Thus, synthesis of xylofuranosyloxymethyl nucleosides was achieved by coupling of xylofuranosyl donor 4 with nucleic acid bases similar to ribofuranosyl donor.[14] It was observed that the formation of glycosyloxymethyl nucleosides was in a higher ratio to natural nucleosides in case of ribofuranosyl donor when compared to xylofuranosyl donor during coupling reactions performed using SnCl4. This may be due to subtle difference in the reactivity of xylose donor to the ribose donor. There is no significant difference observed between ribose and xylose donors when activated by TMSOTf.
CONCLUSION
In summary, we have successfully demonstrated that iterative glycosylation of acetoxymethoxy xylofuranosyl donor resulted in the formation of novel xylofuranosyloxymethyl nucleosides. The coupling reactions using SnCl4 as an activator resulted in the formation of a mixture of natural nucleosides and novel xylofuranosyloxymethyl nucleosides whereas use of TMSOTf resulted in the exclusive formation of novel xylofuranosyloxymethyl nucleosides (in case of uracil, thymine, guanine) and mixture of nucleosides (in case of cytosine and adenine). The application of this new method to other glycosyl donors exploring various Lewis acids and their biological functions will be reported in near future.
EXPERIMENTAL SECTION
General. 1H NMR spectra were recorded using the following instruments: at 200 MHz on a Varian Gemini, at 300 MHz on a Bruker Avance; at 400 MHz on a Varian Unity; at 500 MHz on a Varian Inova, with tetramethyl silane as internal standard for solutions in CDCl3. J values are given Hz. 13C NMR spectra were taken with a Varian Gemini (50 MHz), Bruker Avance (75 MHz) spectrometer with CDCl3 as internal standard (C 77.0) for solutions in deuteriochloroform, DMSO-d6 (C 39.7) for solutions in deuteriodimethyl sulfoxide, dioxane (C 67.3) for solutions in D2O. Optical rotations were measured with a JASCO DIP-370 instrument. Melting points were determined by using Fischer–John’s melting point apparatus and are uncorrected. IR spectra were taken with a Perkin–Elmer 1310 spectrometer. UV spectra were recorded on a Shimadzu UV 160A spectrometer. Organic solutions were dried over anhydrous Na2SO4.
2-Propynyl 2,3,5-tri-O-benzoyl-β-D-xylofuranoside (2)
To a solution of 1-O-acetyl-2,3,5-tri-O-benzoyl-α/β-D-xylofuranoside (1) (30.0 g, 59.5 mmol) in dry CHCl3 (300 mL) was added propargyl alcohol (4.2 mL, 71.4 mmol) and BF3.OEt2 (9 mL, 71.4 mmol) at 0 oC. The reaction mixture was stirred at room temperature for 8 h. After completion of the reaction, anhydrous K2CO3 (9.0 g) was added, stirred for a further 30 min., filtered, and the residue was washed with CHCl3 (75 mL). The filtrate was transferred to a separating funnel, washed with water (2×150 mL) and brine (150 mL). The organic phase was separated, dried (Na2SO4) and concentrated to obtain a residue. The residue was chromatographed [SiO2; 60-120 mesh; hexane-ethyl acetate (9:1)] to isolate the title compound 2 as syrup (19.2 g, 66%). [α]D +10.0o (c 1.0, CHCl3); νmax (KBr)/cm-1 1723; 1H NMR (200 MHz, CDCl3) δ 2.42 (1H, t, CºCH), 4.40 (2H, m, CH2-CºC), 4.50 (1H, dd, J5,5’ =13.0, J4,5 =6.0, H-5), 4.65 (1H, dd, J4,5’ = 4.7, H-5’), 4.96 (1H, 2d, J3,4 = 4.8, H-4), 5.42 (1H, s, H-1), 5.60 (1H, s, H-2), 5.82 (1H d, H-3), 7.20-8.20 (15H, m, Ar-H); 13C NMR (50 MHz, CDCl3) δ 54.6 (HCºC-), 63.4 (C-5), 75.0 (C-4), 75.2 (C-3), 78.6 (O-CH2-C), 79.2 (HCºC-), 81.1 (C-2), 104.1 (C-1), 125-135 (aromatic), 164.9, 165.2 and 166.1 (C=O, ester); Mass (FAB-MS): m/z 523 [M++Na]; Anal. Calcd for C29H24O8: C, 69.59; H, 4.83. Found: C, 69.38; H, 4.80.
2-(Oxopropoxy) 2,3,5-tri-O-benzoyl-β-D-xylofuranoside (3)
To a solution of 2 (19.0 g, 38.0 mmol) in acetone:H2O (300 mL, 2:1) was added Hg(OCOCF3)2 (3.16 g, 7.60 mmol). The reaction mixture was stirred at room temperature for 6 h. After completion of the reaction, acetone was evaporated; the resulting residue was dissolved in ethyl acetate (300 mL), washed with 10% aq. KI solution (2×150 mL), 20% aq. hypo solution (2×150 mL) and brine (150 mL). The organic layer was separated, dried over Na2SO4 and concentrated to obtain the title compound 3 as syrup (18.2 g, 91%). [α]D +18.0o (c 1.0, CHCl3); νmax (KBr)/cm-1 1715 and 1600; 1H NMR (200 MHz, CDCl3) δ 2.25 (3H, s, CH3), 4.20, 4.40 (2H, AB type quartet, Jgem = 15.0, OCH2C), 4.60 (1H, dd, J5,5’ = 13.0, J4,5 = 5.0, H-5), 4.70 (1H, dd, J4,5’ = 6.0, H-5’), 5.00 (1H, 2d, J3,4 = 5.0, H-4), 5.30 (1H, s, H-1), 5.65 (1H, s, H-2), 5.90 (1H, d, H-3), 7.20-8.20 (15H, m, Ar-H); 13C NMR (50 MHz, CDCl3) δ 26.5 (CH3), 63.2 (C-5), 72.5 (C-4), 75.0 (C-3), 79.3 (C-2), 80.9 (-OCH2-), 105.7 (C-1), 125-133 (aromatic), 164.9, 165.2 and 165.9, 205.2 (C=O, ester); Mass (FAB-MS): m/z 541 [M++Na]; Anal. Calcd for C29H26O9: C, 67.17; H, 5.05. Found: C, 66.97; H, 5.16.
1-[(2,3,5-Tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]acetate (4)
To a solution of 3 (17.5g, 33.7 mmol) in dry CHCl3 (175 mL) was added m-CPBA (14.0 g, 81.1 mmol). The reaction mixture was stirred at room temperature for 14 h. After completion of the reaction, the reaction mixture was diluted with CHCl3 (150 mL), washed with saturated aq. NaHCO3 solution (150 mLx3, water (150 mL), brine (150 mL) and dried (Na2SO4). The solvent was concentrated to give a residue which was purified by silica gel chromatography [SiO2; 60-120 mesh; hexane:ethyl acetate (9:1)] to obtain the title compound 4 as a solid (13.4 g, 75%). mp 115-117 oC; [α]D +14.0o (c 1.0, CHCl3); νmax (KBr)/cm-1 1723; 1H NMR (200 MHz, CDCl3) δ 2.10 (3H, s, CH3), 4.58 (1H, dd, J5,5’=13.0, J4,5 = 4.65, H-5), 4.68 (1H, dd, J4,5’ = 6.0, H-5’), 4.98 (1H, 2d, J3,4 = 5.0, H-4), 5.40 – 5.48 (2H, m, OCH2O), 5.50 (1H, s, H-1), 5.55 (1H, s, H-2), 5.88 (1H, d, H-3), 7.20-8.20 (15H, m, Ar-H); 13C NMR (50 MHz, CDCl3) δ 20.9 (CH3) 63.2 (C-5), 74.9 (C-4), 79.5 (C-3), 81.0 (C-2), 85.1 (OCH2O), 104.9 (C-1), 128-135 (aromatic), 164.8, 165.1, 166.0 and 170.2 (C=O, ester); Mass (FAB-MS): m/z 535 [M++H]; Anal. Calcd for C29H26O10: C, 65.16; H, 4.90. Found: C, 65.17; H, 5.14.
A typical procedure for coupling of xylofuranoside donor 4 with nucleic acid bases 5–8, 20, and 24. Method A: SnCl4; Method B: Trimethylsilyltrifluoromethanesulfonate (TMSOTf)
To a solution (20 mL) of xylofuranoside donor 4 (1.0 mmol) and silylated nucleic acid bases 5–8, 20, 24 (1.2 mmol) at 0 oC was added the catalyst (1.2–2.0 mmol, 0.5N in CHCl3 or MeCN or 1,2-dichloroethane) under nitrogen atmosphere. The reaction mixture was stirred at room temperature for 4–18 h or reflux temperature for 1–6 h, progress of the reaction was monitored by TLC. After completion of the reaction it was neutralized with saturated aqueous NaHCO3 solution and filtered through a celite pad. The filtrate was extracted into CHCl3 (30 ml), organic phase was separated, washed with brine, dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to obtain a residue. The residue was separated by column chromatography [SiO2, 60–120 mesh, ethyl acetate:chloroform] to isolate the title compounds.
1-[(2,3,5-Tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]uracil (9)
Compound 9: Mp 83-85 oC; [α]D +17.5o (c 1.0, CHCl3); λmax/nm 259 (MeCN); νmax (KBr)/cm-1 1730; 1H NMR (200 MHz, CDCl3) δ 4.50 (1H, dd, J5’,5” =13.0, J4’,5’ 5.0, H-5’), 4.64 (1H, dd, J4’,5” = 6.0, H-5”), 4.92 (1H, m, H-4’), 5.26, 5.40 (2H, AB type quartet, Jgem = 11.0, OCH2N), 5.44 (1H, s, H-1’), 5.52 (1H, s, H-2’), 5.64 (1H, d, J 3’,4’ = 5.1, H-3’), 5.80 (1H, d, J5,6 = 5.0, H-5), 7.24-8.10 (16H, m, H-6, Ar-H); Mass (FAB-MS): m/z 609 [M++Na]; Anal Calcd for C31H26O10N2: C, 63.48; H, 4.47; N, 4.78 Found: C, 63.26; H, 4.38; N, 4.59. Compound 10: Mp. 108-111 oC [lit.,13 Mp 112-118 oC]; [α]D +73.3o (c 1.0, CHCl3) [lit.,13 [α]D +75.0o (c 0.8, CHCl3)].
1-[(β-D-Xylofuranosyloxy)methyl]uracil (11)
To a solution of 9 (0.8 g, 1.36 mmol) in methanol (15 mL) was added a catalytic amount of NaHCO3 (0.1 g) and refluxed for 3 h. After completion of the reaction the reaction mixture was neutralized with IR 120 H+ resin, filtered, washed with methanol (15 mL) and filtrate was evaporated to a residue. The residue was dissolved in water (30 mL) washed with chloroform (2×25 mL); the water phase was concentrated to isolate the title compound 11 as a thick syrup (0.32 g, 84%). [α]D –55.0o (c 1.0, H2O); λmax/nm 260 (H2O), 261 (0.1N, NaOH); νmax (KBr)/cm-11685; 1H NMR (400 MHz, D2O) δ 3.60 (1H, dd, J5’,5” = 10.0, J4’,5’ = 5.5, H-5’), 3.75 (1H, dd, J4’,5” = 4.7, H-5”), 4.10 (1H, s, H-2’), 4.18 (1H, m, H-4’), 4.30 (1H, m, H-3’), 5.12 (1H, s, H-1’), 5.20, 5.30 (2H, AB type quartet, Jgem = 10.0, OCH2N), 5.80 (1H, d, J5,6 = 5.6, H-5), 7.65 (1H, d, H-6); 13C NMR (50 MHz, D2O) δ 66.0 (C-5’), 81.0 (C-4’), 81.2 (C-3’), 86.3 (C-2’), 89.2 (OCH2N), 108.0 (C-1’), 113.0 (C-5), 152.0 (C-6), 158.0 (C=O,C-2), 172.0 (C=O,C-4); Mass (FAB-MS): m/z 275 [M++H]; Anal. Calcd for C10H14O7N7: C, 43.80; H, 5.15; N, 10.22. Found: C, 43.56; H, 5.22; N, 10.28.
1-[(2,3,5-Tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]thymine (12)
Compound 12: Mp 137-139oC; [α]D +14.2o (c 1.0, CHCl3); λmax/nm 263 (MeOH); νmax (KBr)/cm-1 1723; 1H NMR (200 MHz, CDCl3) δ 1.80 (3H, s, CH3), 4.40-4.70 (2H, m, H-5’,5”), 4.85-5.10 (1H, m, H-4’), 5.22,5.34 (1H, AB type quartet, Jgem = 11.0, OCH2N), 5.40 (1H, s, H-1’), 5.52 (1H, s, H-2’), 5.80 (1H, d, J3’,4’ = 5.4, H-3’), 7.10 (1H, s, H-6), 720-8.10 (15H, m, Ar-H), 9.05 (1H, br s, NH); 13C NMR (50 MHz, CDCl3) δ 12.2 (CH3), 63.5 (C-5’), 74.5 (C-3’), 75.0 (C-2’), 79.5 (C-4’), 81.0 (OCH2N), 105.0 (C-1’), 111.5 (C-5), 125-135 (aromatic), 139.1 (C-6), 150.5 (C=O,C-2) 164.0 (C=O,C-4) 164.5, 165.0, 166.0 (C=O, ester); Mass (FAB-MS): m/z 601 [M++H]; Anal. Calcd for C32H28O18N2: C, 63.99; H, 4.70; N, 4.66. Found: C, 63.28; H, 4.62; N, 4.73.
Compound 13: Mp 196-199 oC [lit.14 mp. 196-198 oC]; [α]D +49.3o (c 0.85, CHCl3) [lit.14 [α]D +50.6o (c 0.85, CHCl3)].
1-[(β-D-Xylofuranosyloxy)methyl]thymine (14)
The benzoylated derivative 12 (0.8g, 1.33 mmol) in methanol (16 mL) was deprotected as described for 11 to isolate the title compound 14 as a white solid (0.35 g, 91.0%). mp 175-177 oC; [α]D –31.0o (c 1.0, H2O); λmax/nm 265 (H2O), 266 (0.1N NaOH); νmax (KBr)/cm-1 1690; 1H NMR (200 MHz; D2O) δ 1.88 (3H, s, CH3), 3.70 (1H, dd, J5’,5” = 14.0, J4’,5’ =5.2, H-5’), 3.85 (1H, dd, J4’,5” = 4.0, H-5”), 4.15 (1H, d, J2’,3’ = 4.65, H-3’), 4.26 (1H, d, H-2’), 4.30-4.45 (1H, m, H-4’), 5.18 (1H, s, H-1’), 5.20, 5.35 (2H, AB type quartet, Jgem = 14.0, OCH2N), 7.45 (1H, s, H-6), 8.00 (1H, br s, NH); 13C NMR (50 MHz, D2O) δ 12.5 (CH3), 60.3 (C-5’), 73.9 (C-4’), 75.5 (C-3’), 80.5 (C-2’), 83.2 (OCH2N), 106.5 (C-1’), 109.0 (C-5), 140.0 (C-6), 154.0 (C=O,C-2), 168.0 (C=O,C-4); Mass (FAB-MS): m/z 311 [M++Na]; Anal. Calcd for C11H16O7N2: C, 45.83, H, 5.60, N, 9.2. Found: C, 45.67; H, 5.48; N, 8.91.
1-[(2,3,5-Tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]cytosine (15)
Compound 15: Mp 122-126oC; [α]D +44.6o (c 1.0, CHCl3); λmax/nm 274 (MeOH); νmax (KBr)/cm-1 1723, 1661 and 1615; 1H NMR (200 MHz, CDCl3) δ 4.48 (1H, dd, J5’,5” = 13.0, J4’,5’ = 5.0, H-5’), 4.62 (1H, dd, J4’,5” =6.0, H-5”), 4.90 (1H, dd, J3’,4’ = J4’,5’ 5.20, H-4’), 5.25, 5.35 (2H, AB type quartet, Jgem = 9.6, OCH2N), 5.48 (1H, s, H-1’), 5.50 (1H, s, H-2’), 5.76 (1H, d, H-3’), 5.82 (1H, d, J5,6 = 7.6, H-5), 7.20-7.60 (10H, m, H-6, Ar-H), 7.85-8.20 (6H, m, Ar-H); 13C NMR (50 MHz, CDCl3) δ 63.3 (C-5’), 75.1 (C-4’), 76.3 (C-3’), 79.4 (C-2’), 81.0 (OCH2N), 95.6 (C-5), 104.8 (C-1’), 128.0-135.0 (aromatic), 144.7 (C-6), 155.9 (C-4), 165.0 (C=2, C=O), (C=O,C-2) 165.6, 166.0, 166.2 (C=O, ester); Mass (FAB-MS): m/z 586 [M++H]; Anal. Cacld for C31H27O9N3: C, 63.58; H, 4.65; N, 7.18. Found: C, 63.32; H, 4.63; N, 7.11.
Compound 16: Mp 137-139 oC [lit.,14 mp 140-141 oC]; [α]D +61.5o (c 0.6, CHCl3) [lit.,14 [α]D +63.4o (c 0.6, CHCl3)].
1-[(β-D-Xylofuranosyloxy)methyl]cytosine (17)
The benzoylated derivative 15 (0.6 g, 0.9 mmol) was debenzoylated as described for 14 to isolate the title compound 17 as a syrup (0.19 g, 81.5%). [α]D +17.8o (c 1, H2O); λmax/nm 268 (H2O), 270 nm (0.1N NaOH); νmax (KBr)/cm-1 1653; 1H NMR (200 MHz, D2O) δ 3.70 (1H, dd, J5’,5” = 12.8, J4’,5’ = 5.0, H-5’), 3.84 (1H, dd, J4’,5” = 6.0, H-5”), 4.20 (1H, s, H-2’), 4.22-4.40 (2H, m, H-3’, H-4’), 5.20 (1H, s, H-1’), 5.24, 5.40 (2H, AB type quartet, Jgem = 10.0, OCH2N), 6.20 (1H,d, J5,6 = 7.8, H-5), 7.70 (1H, d, H-6); 13C NMR (50 MHz, D2O) δ 59.3 (C-5’), 73.5 (C-4’), 74.9 (C-3’), 78.7 (C-2’), 81.6 (OCH2N), 104.9 (C-1’), 94.6 (C-5), 144.8 (C-6), 156.0 (C=O, C-2), 165.2 (C=O,C-4); Mass (FAB-MS) m/z 274 [M++H]; Anal. Calcd for C10H15O6N2: C, 43.95; H, 5.53; N, 15.38. Found: C, 44.11; H, 5.38; N, 15.21.
N6-Benzoyl-9-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]adenine (21)
Compound 21: Mp 77-79 oC; [α]D +32.6o (c 1, CHCl3); λmax/nm 277 (MeCN); νmax (KBr)/cm-1 1723 and 1600. 1H NMR (200 MHz, CDCl3) δ 4.50 (2H, m, H-5’,5”), 4.98 (1H, m, H-4’), 5.50 (1H, s, H-1’), 5.56 (1H, s, H-2’), 5.80 (1H, d, J3’,4’ = 3.5, H-3’), 5.84, 6.02 (2H, AB type quartet, Jgem = 9.8, OCH2N), 7.20-7.60 (12H, m, Ar-H), 7.80-8.16 (8H, m, Ar-H), 8.22 (1H, s, H-8), 8.78 (1H, s, H-2), 9.24 (1H, br s, NH); Mass (FAB-MS): m/z 714 [M++H]; Anal. Cacld for C39H31O9N5: C, 65.68; H, 4.37; N, 9.82. Found: C, 63.23; H, 4.48; N, 9.38. Compound 22: Mp 104-107 oC [lit.,14,17 mp 105-110 oC]; [α]D +10.73o (c 1.0, CHCl3) [lit.,14,17 [α]D +5.2o (c 0.8, CHCl3)];
9-[(β-D-Xylofuranosyloxy)methyl]adenine (23)
The benzoylated derivative 21 (0.6 g, 0.84 mmol) was debenzoylated as described for 11 to isolate the title compound 23 as a solid (0.19 g, 76.0%). mp 220 oC (decom.); [α]D –38.5o (c 1, DMSO); λmax/nm 259 (H2O); νmax (KBr)/cm-1 1678 and 1610; 1H NMR (300 MHz, DMSO-d6) δ 3.20-4.30 (6H, m, H-2’,H-3’,H-4’,H-5’,H-5”,OH), 4.82 (1H, br s, OH), 4.98 (1H, s, H-1’), 5.15 (1H, br s, OH), 5.64, 5.85 (2H, AB type quartet, Jgem = 10.0, OCH2N)), 7.30 (2H, br s, NH2), 8.28 (1H, s, H-8), 8.30 (1H,s, H-2); 13C NMR (50 MHz, DMSO-d6) δ 63.1 (C-5’), 68.8 (C-4’), 71.8 (C-3’), 74.4 (C-2’), 86.1 (OCH2N), 105.5 (C-1’), 119.3 (C-5), 143.1 (C-8), 149.8 (C-4), 153.6 (C-2), 156.0 (C-6); Mass (FAB-MS): m/z 298 [M++H]; Anal. Calcd for C11H15O5N5: C, 44.44; H, 5.09; N, 23.56. Found: C, 43.91; H, 5.14; N, 23.42.
N2-Acetyl-9-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]guanine (25) and N2-acetyl-7-[(2,3,5-tri-O-benzoyl-β-D-xylofuranosyloxy)methyl]-guanine (26)
Compound 25: Mp 149-151 oC; [α]D –4.89o (c 1.0, CHCl3); λmax/nm 276 (MeCN); νmax (KBr)/cm-1 1715, 1615 and 1570; 1H NMR (200 MHz, CDCl3) δ 2.20 (3H, s, OCOCH3), 4.60 (1H, dd, J5’,5” =12.8, J4’,5’ = 6.0, H-5’), 4.90-5.20 (2H, m, H-4’, H-5), 5.30, 5.78 (2H, AB type quartet, Jgem = 10.0, OCH2N), 5.50 (1H, s, H-1’), 5.54 (1H, s, H-2’), 5.84 (1H, d, J3’,4’ = 6.5, H-3’), 7.08-7.70 (10H, H-8, Ar-H), 8.00 (6H, d, Ar-H), 10.49 (1H, br s, NH), 11.90 (1H, br s, NH); Mass (FAB-MS): m/z 668 [M++H]; Anal. Calcd for C34H29O10N5: C, 61.16; H, 4.38; N, 10.49. Found: C, 61.08; H, 4.12; N, 9.98.
Compound 26: Mp 115-120 oC; [α]D +2.7o (c 1.0, CHCl3); λmax/nm 261 (MeCN); νmax (KBr)/cm-1 1715, 1615 and 1570; 1H NMR (200 MHz, CDCl3) δ 2.40 (3H, s, OCOCH3), 4.38-4.52 (2H, m, H-4’, H-5”), 4.94 (1H, m, H-4’), 5.58 (1H, s, H-1’), 5.62 (1H, s, H-2’), 5.80 (d, J3’,4’ 6.3, H-3’), 5.98, 6.05 (2H, AB type quartet, Jgem = 10.0, OCH2N), 7.30-7.64 (9H, m, Ar-H), 7.92-8.10 (7H, m, H-8, Ar-H), 11.10 (1H, br s, NH), 12.30 (1H, br s, NH); Mass (FAB-MS): m/z 668 [M++H]; Anal. Cacld for C34H29O10N5: C, 66.16; H, 4.38; N, 10.49. Found: C, 66.21; H, 4.23; N, 10.23.
9-[(β-D-Xylofuranosyloxy)methyl]guanine (29)
The benzoyl derivative 25 (0.4g, 0.59 mmol) in methanol (10 mL) was deprotected as described for 11 to isolate the title compound 29 as a solid (0.14 g, 78%). mp 201-203 oC; [α]D –43.4o (c 1.0, DMSO); λmax/nm 251 (H2O); νmax (KBr)/cm-1 3400, 1723, 1685 and 1623; 1H NMR (200 MHz, DMSO-d6) δ 3.40-3.70 (2H, m, H-5’, H-5”), 3.80 (1H, m, H-2’), 3.90 (1H, m, H-3’), 4.10 (1H, dd, J3’,4’ =3.5, J4’,5’ = 6.0, H-4’), 4.50 (1H, s, OH), 4.80 (2H, br s, 2xOH), 5.30 (1H, s, H-1’), 5.36, 5.46 (2H, AB type quartet, Jgem = 9.8, OCH2N), 6.58 (2H, br s, NH2), 7.78 (1H, s, H-8), 10.68 (1H, br s, NH); 13C NMR (50 MHz, DMSO-d6) δ 60.5 (C-5’), 68.3 (C-4’), 75.5 (C-3’), 80.9 (C-2’), 83.3 (OCH2N), 105.9 (C-1’), 116.4 (C-5), 137.7 (C-8), 151.3 (C-4), 153.8 (C-2), 156.7 (C-6); Mass (FAB-MS): m/z 314 [M++H]; Anal. Calcd for C11H15O6N5: C, 42.17; H, 4.83; N, 22.36. Found: C, 42.31; H, 4.67; N, 23.43.
7-[(β-D-Xylofuranosyloxy)methyl]guanine (30)
The benzoylated derivative 26 (0.2 g, 0.29 mmol) in methanol (6 mL) was deprotected as described for 11 to isolate the title compound 30 as a white solid (0.07 g, 75.0%). Mp 235 oC (decom.); [α]D –47.3 (c 0.5, DMSO); λmax/nm 281 (H2O); νmax (KBr)/cm-1 1677 and 1485; 1H; NMR (200 MHz, DMSO-d6) δ 3.70-4.20 (5H, m, H-2’, 3’, 4’, 5’, 5”), 4.60 (1H, s, OH), 4.80 (1H, s, OH), 4.95 (1H, s, H-1’), 5.00 (1H, s, OH), 5.62, 5.70 (2H, AB type quartet, Jgem = 8.8, OCH2N), 6.70 (2H, br s, NH2), 8.30 (1H, s, H-8), 10.90 (1H, br s, NH); 13C NMR (75 MHz, DMSO-d6) δ 60.5 (C-5’), 72.6 (C-4’), 75.4 (C-3’), 80.9 (C-2’), 83.6 (OCH2N), 106.2 (C-1’), 106.7 (C-5), 143.2 (C-8), 154.1 (C-2), 155.2 (C-6), 160.8 (C-4); Mass (FAB-MS): m/z 314 [M++H]; Anal. Calcd for C11H15O6N5: C, 42.17; H, 4.83; N, 22.36. Found: C, 42.25; H, 4.41; N, 22.62.
ACKNOWLEDGEMENTS
The authors are grateful to the Indian Institute of Chemical Technology, Hyderabad and thankful to Council of Scientific and Industrial Research, New Delhi for the financial support.
REFERENCES
- (a) Pfleiderer, W. Pyrimidine — Purine – Pteridine: An Introduction to the Chemistry and Biochemistry of Pyrimidines, Purines, and Pteridines. Von D. T. Hurst. John Wiley, Chichester – New York – Brisbane – Toronto, VIII: 266S (1979). (b) Blackburn, G.M., Gait, M.J. Nucleic Acids in Chemistry and Biology; Ed.; IRL: New York, (1992).
- Ichikawa E., Kato K. Sugar-modified nucleosides in past 10 Years, A review. Curr. Med. Chem., 8: 385-423 (2001).
- Huryn, D. M., Okabe, M. AIDS driven nucleoside chemistry Chem. Rev., 92: 1745-1708 (1992).
- Matsuda, A., Sasaki, T. Antitumor activity of sugar-modified cytosine Nucleosides. Cancer Sci., 95: 105-111 (2004).
- Ohrui, H. Development of modified nucleosides that have supremely high anti-HIV activity and low toxicity and prevent the emergence of resistant HIV mutants. Proc. Jpn. Acad., Ser. B, 87: 53-65 (2011).
- Gao, H., Mitra, A.K. Synthesis of acyclovir, ganciclovir and their prodrugs: A review. Synthesis, 329-351 (2000).
- (a) Mydock, L.K., Demchenko, A. V. Mechanism of chemical O-glycosylation: From early studies to recent discoveries. Org. Biomol. Chem., 8: 497-510 (2010). (b) Zhu, X., Schmidt, R.R. New principles for glycoside-bond formation. Angew. Chem. Int. Ed. Engl., 48: 1900-1934 (2009). (c) Toshima, K.; Tatsuta, K. Recent progress in O-glycosylation methods and its application to natural products synthesis. Chem. Rev., 93: 1503-1531 (1993).
- (a) Rencurosi, A., Poletti, L., Russo, G., Lay, L. Lipase-catalysed regioselective acylations in combination with regioselective glycosylations as a strategy for the synthesis of oligosaccharides: Synthesis of a series of fucosyllactose building blocks. Eur. J. Org. Chem., 1672-1680 (2003). (b) Zeng, Y., Kong, F. Regioselective glycosylation of 4,6-O-benzylidenated glucopyranosides. Carbohydr. Res., 338: 843-849 (2003).
- (a) Geurstsen, R., Boons, G. J. Regioselective glycosylations in solution and on soluble and insoluble polymeric supports. Eur. J. Org. Chem., 1473-1477 (2002). (b) Li, B., Yu, B., Hui, Y., Li, M., Han, X., Fung, K.-P. An improved synthesis of the saponin, polyphyllin D. Carbohydr. Res., 331: 1-7 (2001). (c) Ellervick, U., Magnusson, G. A high yielding chemical synthesis of sialyl lewis x tetrasaccharide and lewis x trisaccharide; Examples of regio and stereodifferentiated glycosylations. J. Org. Chem., 63: 9314-9322 (1998). (d) Demchenko, A.; Boons, G.J. A. Highly convergent synthesis of a hexasaccharide derived from the oligosaccharide of group B Type III streptococcus. Tetrahedron Lett. 38: 1629-1632 (1997).
- (a) Boons, G.J. Strategies in oligosaccharide synthesis. Tetrahedron, 52: 1095-1121 (1996). (b) Schmidt, R.R., Kinzy, W. Anomeric-oxygen activation for glycoside synthesis: The trichloroacetimidate method. Adv. Carbohydr. Chem. Biochem. 50: 21-123 (1994).
- (a) Wolfe, S., Whangbo, M.H., Mitchell, D. J. On the magnitudes and origins of the “anomeric effects”, “exo-anomeric effects”, “reverse anomeric effects”, and c-x and c-y bond lengths in XCH2YH molecules. Carbohydr. Res., 69: 1-26 (1979) (b) Tvaroska, I., Bleha, T. Anomeric and exo-anomeric effects in carbohydrate chemistry. Adv. Carbohydr. Chem. Biochem., 47: 45-123 (1989) (c) Box, V.G.S. The role of lone pair interactions in the chemistry of the monosaccharides. The anomeric effect. Heterocycles, 31: 1157-1181 (1990).
- (a) Mereyala, H.B., Gurrala, S.R., Gadikota, R.R. Stereoselective synthesis of α-O-fucosylated serine, threonine, tyrosine and saccharides. J. Chem. Soc., Perkin Trans. 1: 3179-3182 (1997). (b) Mereyala, H.B., Gurijala, V.R. Use of 2-pyridyl 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-1-thio-β-D-glucopyranoside as a glycosyl donor and methyl iodide as an activator for the synthesis of 1,2-trans linked saccharides. Carbohydr. Res., 242: 277-280 (1993).
- Mereyala, H.B., Gurrala, S.R. Design, Development and utility of glycosyl donors bearing an acetoxymethoxy leaving group. Chem. Lett. 863-864 (1998).
- Mereyala, H.B., Mamidyala, S.K. The synthesis of [(β-D-ribofuranosyloxy)-methyl] nucleosides. Nucleosides Nucleotides & Nucleic Acids, 23: 655-669 (2004).
- Mereyala, H.B., Gurrala, S.R. A highly diastereoselective, practical synthesis of allyl, propargyl 2,3,4,6-tetra-O-acetyl-β-D-gluco, β-D-galactopyranosides and allyl, propargyl heptaacetyl-β-D-lactosides. Carbohydr. Res., 307: 351-354 (1998).
- (a) Nakayama, C., Saneyoshi, M. Synthetic nucleosides and nucleotides. XX. Synthesis of various 1-β-D-xylofuranosyl-5-alkyluracils and related nucleosides. Nucleosides, Nucleotides, 1: 139-146 (1982). (b) Niedballa, U., Vorbruggen, H. A general synthesis of N-glycosides. 6. On the mechanism of the stannic chloride catalyzed silyl Hilbert-Johnson reaction. J. Org. Chem., 41: 2084-2086 (1976).
- (a) Codington, J.F., Fecher, R., Fox, J.J. Pyrimidine Nucleosides. VII. Reactions of 2’,3’,5’-trimesyloxyuridine. J. Am. Chem. Soc., 82: 2794-2803 (1960).
- (a) Fox, J.J., Codington, J.F., Yung, N.C., Kaplan, L., Lampen, J.O. Pyrimidine nucleosides. IV. The synthesis of 1-β-D-lyxofuranosylthymine. J. Am. Chem. Soc., 80: 5155-5160 (1958). (b) Gosselin, G., Bergogne, M.-C., de Rudder, J., De Clercq, E., Imbach, J.-L. Systematic synthesis and biological evaluation of α- and β-D-xylofuranosyl nucleosides of the five naturally occurring bases in nucleic acids and related analogues. J. Med. Chem., 29: 203-213 (1986). (c) Shugar, D., Fox, J.J. Spectrophotometric studies of nucleic acid derivatives and related compounds as a function of pH. I. Pyrimidines. Biochem. Biophys. Acta., 9: 199-218 (1952).
- (a) Baizer, M.M., Clark, J.R., Dub, M., Loter, A. A new synthesis of kinetin and its analogs. J. Org. Chem., 21: 1276-1277 (1956).
- Miyaki, M., Shimizu, B. N N and Glycosyl migration of purines and pyrimidines. III. N N Alkyl migration of purine derivatives. Chem. Pharm. Bull., 18: 1446-1456 (1970).
- (a) Pugmire, R.J., Grant, D.M., Robins, R.K., Rhodes, G.W. Carbon-13 magnetic resonance. III. Purine. J. Am. Chem. Soc., 87: 2225-2228 (1965). (b) Garner, P.; Ramakanth, S. A regiocontrolled synthesis of N7– and N9-guanine nucleosides. J. Org. Chem. 53: 1294-1298 (1988). (c) Kjellberg, J.; Johansson, N. G. Characterization of N7 and N9 alkylated purine analogues by 1H and 13C NMR. Tetrahedron, 42: 6541-6544 (1986).







ISSN Online: 2231-5039

![]()




{kind=link}