Synthesis of 4-propionyl-3-(4-substitutedphenylamino)- 2-(5-Nitropyridin-2-yl) isoxazol-5(2H)-ones and Their Rearrangements to Imidazo [1,2- a] pyridines and Indoles with Triethylamine (TEA)
Chalak Azimi* and Snoor Majidi
Department of Chemistry Management, College of Chemistry, Piranshahr Branch, Islamic Azad University, Piranshahr, Iran
DOI : http://dx.doi.org/10.13005/ojc/300229
Article Received on :
Article Accepted on :
Article Published : 21 May 2014
4-propionyl-3-(4-Substitutedphenylamino)isoxazol-5(2H)-ones, substituted on nitrogen with a 2-chloro-5-nitropyridine group, react with triethylamine (TEA) to give imidazo [1,2-a]pyridines and indoles. With 4-bromophenyl and 4-methylphenyl group substituents only imidazopyridines are formed, but the 4-methoxyphenyl derivative gave a 3:1 mixture of the corresponding imidazo[1,2- a]pyridine and 2-pyridylaminoindole, respectively.
KEYWORDS:Isoxazolones; Triethylamine (TEA); Imidazopyridines; Indoles
Download this article as:| Copy the following to cite this article: Azimi C, Majidi S. Synthesis of 4-propionyl-3-(4-substitutedphenylamino)- 2-(5-Nitropyridin-2-yl) isoxazol-5(2H)-ones and Their Rearrangements to Imidazo [1,2- a] pyridines and Indoles with Triethylamine (TEA). Orient J Chem 2014;30(2). |
| Copy the following to cite this URL: Azimi C, Majidi S. Synthesis of 4-propionyl-3-(4-substitutedphenylamino)- 2-(5-Nitropyridin-2-yl) isoxazol-5(2H)-ones and Their Rearrangements to Imidazo [1,2- a] pyridines and Indoles with Triethylamine (TEA). Orient J Chem 2014;30(2) Available from: http://www.orientjchem.org/?p=3355 |
Introduction
Khalafy et al. recently reported1 that the reaction of 2-aryl-3-phenylaminoisoxazolones 1, substituted on nitrogen with an isoquinoline or quinazoline group, react with triethylamine to give Imidazo annelated compounds 2 and 3 respectively (Scheme 1). When the N-substituent is a nitropyridine, the 2-aminoindole structure 4 was assigned to the product. Evidence was presented that the reactions proceed by initial addition of the tertiary amine to C-4. In this paper we detail further research that suggests that both the proposed structure for 4, and the reaction pathway, require modification.
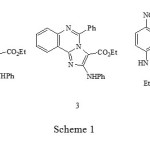 |
Scheme 1 |
These results are formally the same as those achieved by photolysis or pyrolysis of the corresponding N-substituted isoxazolones2. However, the reaction of 3-subtituted isoxazolones with bases is not so well known, and the only examples appear to be those reported by Doleschall 3.
We have also reported4 rearrangement of 4-acetyl-3-(4-substituted phenylamino)-2-(5-nitropyridin-2-yl) isoxazol-5(2H)-ones (6, X: Br, Me, OMe) to Imidazo [1, 2-a]pyridines (7, X: Br, Me, OMe) under Flash-Vacuum-Pyrolysis (F.V.P) conditions (Scheme 2).
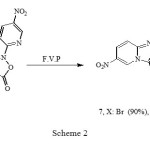 |
Scheme 2 |
Here we describe the synthesis of new of 4-propionyl-3-(4-Substitutedphenylamino)isoxazol-5(2H)-ones (11a–c), where the N-substituent is a 2-chloro-5-nitropyridine group, and their reactions with triethylamine to form either imidazopyridines or indoles.
Results and Discussion
The 2-unsubstituted isoxazolones 10a–c were prepared by the general method of Worral 5. Thus, the reaction of the sodium salt of ethyl-3-oxopentanoate in ethanol with various aryl isothiocyanates gave the thiocarbamates 8a–c in high yield (Scheme 3).
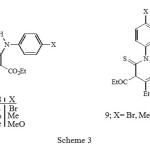 |
Scheme 3 Click here to View Scheme |
The appearance of two different quartets for the ethoxy groups and two different carbonyl groups in the 1H- NMR (500 MHz) and FT-IR spectra of carbamates 8a-c is due to their non equivalency arising from strong H-bonding (see 7), resulting in H-bonded and free ester groups. Such H-bonding has also been deduced from a study of their infrared spectra 6 and acidity 7. The reaction of these carbamates 8a-c with three equivalent of hydroxylamine gave the corresponding isoxazolones 10a- c in good yield (Scheme 4).
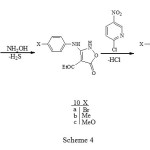 |
Scheme 4 Click here to View Scheme |
Refluxing the isoxazolone 10b with one equivalent of 2-chloro-5-nitropyridine in butanol for 12 hours gave the butyl ester analogue 12 by acid catalysed transesterification (Scheme 5); reaction in the absence of solvent gave the ethyl esters 11.
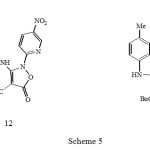 |
Scheme 5 Click here to View Scheme |
N-Substituted isoxazolones 11a and 11b reacted with triethylamine in refluxing ethanol to give the corresponding imidazo[1,2-a]pyridines 13a and 13b as the only products in 84% and 75% yield respectively, but the isoxazolone 11c gave the corresponding imidazo compound 13c as a
major product (59%) with a significant amount of a second product (20%), whose spectral properties were more consistent with those expected for the indole 14. The imidazopyridine structures of 13a-c could clearly be deduced from the similarity of the coupling pattern for the protons in the 4-substituted phenyl ring to that in the starting materials 11a-c, and the indole structure 14 had proton coupling similar to those of the nitropyridyl ring in 11c. The 1H-NMR spectrum of compound 13a showed a doublet of doublets at d 8.19 ppm with J1=9.7Hz and J2=1.3Hz due to H-7, which collapsed to a doublet with J=9.7Hz by irradiation of a broad doublet with J=1.3Hz at d 9.87 ppm due to H-5. However, the 1H-NMR spectra of compounds 13b, 13c and 13 showed H-7 to have meta coupling with H-5, but in none could the resonance for H-5 be clearly observed. The reason for the extreme broadening of this peak is unknown, though quadrupole coupling with N-4 is suspected. Finally, the rearrangement of the isoxazolone 12 with triethylamine in refluxing ethanol gave the corresponding imidazopyridine 13 in 81% yield. The reaction pathway resulting in the imidazopyridines, consistent with our earlier suggestion 1, is shown in Scheme 7.
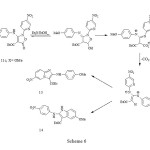 |
Scheme 6 Click here to View Scheme |
While it is possible that the steric effect of the substituent at C-4 of the phenylamino group in the zwitterionic intermediate in Scheme 7 could affect the mode of cyclisation, the differences are more likely to have an electronic origin. We have found that the 4-methoxy derivative 11c reacts rapidly in refluxing ethanol (ca. 15 minutes, compared with 3 h for the corresponding reaction with triethylamine) to form a mixture of imidazopyridine and indole in a 2:1 ratio, respectively. Since the triethylamino group would be unlikely to retain a positive charge under the basic conditions, and thus would be unlikely to act as a leaving group, we feel that Scheme 7 is no longer tenable. An alternative, which is consistent with the electronic requirements of the reaction, is shown in Scheme 6.
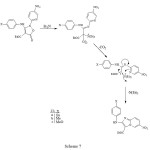 |
Scheme 7 Click here to View Scheme |
These rearrangements, therefore, appear to be generally applicable to the synthesis of imidazo heterocycles and indoles, which are suitable synthetic intermediates for a series of polycyclic heterocycles with possible pharmaceutical applications 8,9.
Conclusion
In conclusion we have shown that a variety of 4-propionyl-3-(4-substituted phenylamino)-2-(5-nitropyridin-2-yl) isoxazol-5(2H)-ones, rearranged with Triethylamineto give imidazo [1,2-a] pyridines and Indoles. These rearrangements, therefore, appear to be generally applicable to the synthesis of imidazoheterocycles which are suitable synthetic intermediates for a series of polycyclic heterocycles with possible pharmaceutical applications.
Experimental Section
General
Freshly distilled solvents were used throughout, and anhydrous solvents were dried according to Perrin and Amarego.10 1H-NMR and 13C-NMR spectra were recorded, in deuteriochloroform, unless otherwise stated, at 500 and 125 MHz respectively, with a Bruker DRX-500 Avance spectrometer. Tetramethylsilane was used as an internal standard and all signals due to amino protons were removed by exchange with D2O. Infrared spectra were recorded on a Unicam Matsson 1000 Fourier-Transform Spectrometer. Mass spectra were recorded on a Varian Matt 311 spectrometer and relative abundance of fragments are quoted in parentheses after the m/z values. Melting points were determined on a Philip Harris C4954718 apparatus and are uncorrected. Micronalyses were preformed on a Carlo–Erba Analyzer 1104.
Ethyl-2-(4-bromophenyl) carbamothioyl-3-oxopentanoate (8a)
Sodium (2.9 g, 0.126 mol) was reacted with absolute ethanol (50 mL) and ethyl-3-oxopentanoate (18.14g, 18 ml, 0.126 mol) was added at rt. The reaction mixture was stirred at room temperature for 15 min, 4–bromophenyl isothiocyanate (26.96 g, 0.126 mol) was added and stirring was continued for further 2 h. The resulting precipitate was filtered off and washed with light petroleum to give the salt of 8a (36.91g, 80%) as yellow crystals, m.p.=161-162 ºC. The salt was dissolved in water (30-40 mL) and neutralized by dropwise addition of dilute HCl. The mixture was stirred for 15 min and the precipitate was filtered to give the title compound (26.02g, 77%) as a pale yellow solid, m.p.= 53-54ºC
1H-NMR(CDCl3)(δppm): 1.06 (t, J=7.1Hz, 3H), 1.3 (t, J=7.1Hz, 3H), 2.5 (q, J=7.1Hz, 2H), 4.32 (q, J=7.1Hz, 2H), 5.09 (s, 1H), 7.55 (d, J=8.6Hz, 2H), 7.73 (d, J=8.6Hz, 2H), 10.9 (bs, 1H, NH).
13C-NMR(CDCl3)(δppm): 8.2, 14.34, 27.8, 63.63, 70.68, 120.37, 125.13, 132.37, 137.89, 166.08, 188.05.
FT-IR: = 3285, 1759, 1723, 1548, 1431, 1285, 1146, 1023, 831 cm-1.
The following thiocarbamates were made by the same procedure.
Ethyl-2-(4-methylphenyl) carbamothioyl-3-oxopentanoate (8b).
Pale yellow solid (90%); m.p.= 55-56 ºC)
1H-NMR(CDCl3)(δppm): 1.06 (t, J=7.1Hz, 3H), 1.3 (t, J=7.1Hz, 3H), 2.35 (s, J=7.1Hz, 3H), 2.49 (q, J=7.1Hz, 2H), 4.32 (q, J=7.1Hz, 2H), 5.09 (s, 1H), 7.23 (d, J=8.3Hz, 2H), 7.66 (d, J=8.3Hz, 2H), 10.77 (bs, 1H, NH).
13C-NMR(CDCl3)(δppm): 8.2, 14.35, 23.57, 34.3 63.47, 67.62, 123.64, 129.86, 136.41, 137.43, 166.16, 187.68.
FT-IR = 3284,1760, 1723, 1515, 1430, 1315, 1223, 1148, 1020, 831 cm-1.
Ethyl-2-(4-methoxyphenyl) carbamothioyl-3-oxopentanoate (8c).
Pale yellow solid (87%); m.p.= 58-59 ºC
1H-NMR(CDCl3)(δppm): 1.06 (t, J=7.1Hz, 3H), 1.3 (t, J=7.1Hz, 3H), 2.5 (q, J=7.1Hz, 2H), 3.73 (s, OMe, 3H), 4.312 (q, J=7.15 Hz, 2H), 5.10 (s, 1H), 6.94 (d, J=8.9Hz, 2H), 7.67 (d, J=8.9Hz, 2H), 10.7 (bs, 1H, NH).
13C-NMR(CDCl3)(δppm): 8.2, 14.1, 34.47, 55.87, 63.46, 76.38, 114.39, 125.29, 131.94, 158.62, 166.18, 187.55.
FT-IR =3284, 1760, 1723, 1515, 1306,1254, 1151, 1030, 842 cm-1.
4-propionyl-3-(4-bromophenylamino)isoxazole-5(2H)-one (10a).
To a solution of hydroxylamine hydrochloride (7.06 g, 102 mmol) in water (30 mL), sodium bicarbonate (10.17 g, 102 mmol) was added slowly. Ethanol (80 mL) was added and the resulting potassium chloride was filtered off. Ethyl-2-(4-bromophenyl) carbamothioyl-3-oxopentanoate (6a, 12.13g, 34 mmol) was added to the filtrate and the mixture was stirred at room temperature for 24 h. The reaction mixture was acidified with dilute HCl and the white precipitate was collected and recrystallized from acetone to give the title product (8.78 g, 79%) as colourless needles, m.p.= 201 º C (dec.).
1H-NMR (d6-DMSO)(δppm): 1.11 (t, J=7.1Hz, 3H), 3.01 (q, J=7.1Hz, 2H), 7.37(d, J=8.4Hz, 2H), 7.57 (d, J=8.4Hz, 2H), 8.30 (bs, 1H, NH), 9.39 (bs, 1H, NH).
13C-NMR (d6-DMSO)(δppm): 7.9, 30.7, 59.96, 84, 118.02, 132.94, 137.10, 163.53, 164.74, 167.39.
FT-IR = 3250, 2950, 2740, 1723, 1696, 1666, 1607, 1563, 1456, 1398, 1316, 1183, 1018, 818 cm-1.
The following compounds were made by the same procedure
4-propionyl-3-(4-methylphenylamino)isoxazole-5(2H)-one(10b).
Refluxing for 24 h gave colourless crystals (85%), m.p.= 165 -167 ºC (dec.).
1H-NMR (d6-DMSO + CDCl3)(δppm): 1.11 (t, J=7.1Hz, 3H), 2.35 (s, Me, 3H), 3.01 (q, J=7.1Hz, 2H), 6.78 (d, J=9.2Hz, 2H), 6.79(bs, 1H, NH), 6.80(d, J=9.2Hz, 2H), 8.85 (bs, 1H, NH).
13C-NMR (d6-DMSO+ CDCl3)(δppm): 7.9, 24.52, 30.85, 84.2, 121.53, 130.13, 133.29, 135.64, 163.59, 165.51, 166.74.
FT-IR = 3669, 2979, 2746, 1705, 1669, 1615, 1331, 1208, 1115, 1023, 800 cm-1.
4-propionyl-3-(4-methoxyphenylamino)isoxazole-5(2H)-one (10c).
Refluxing for 24 h gave the desired product (7.75g, 80%) which was recrystallized from ethanol/acetone (1:1) as a white solid, m.p.= 207-208 ºC (dec.).
1H-NMR (d6-DMSO+CDCl3)(δppm): 1.11 (t, J=7.1Hz, 3H), 3.01 (q, J=7.1Hz, 2H), 3.35 (s, OMe, 3H), 6.38 (d, J=8.5Hz, 2H), 6.94 (d, J=8.5Hz, 2H), 6.96 (bs,1H, NH), 7.70 (bs, 1H, NH).
13C-NMR(d6-DMSO+CDCl3)(δppm): 7.9, 30.90, 55.55, 73.5, 114.6, 118.6, 135.73, 153.7, 165.73, 168.14, 174.81.
FT-IR = 3407, 1708, 1615, 1554, 1248, 1077, 792 cm-1.
4-propionyl-3-(4-bromophenylamino)-2-(5-nitropyridin-2-yl)isoxazole-5(2H)-one (11a).
A mixture of 2-chloro-5-nitropyridine (48.5 mg, 0.30 mmol) and 4-propionyl- 3-(4-bromophenylamino)isoxazole-5(2H)-one (8a, 93 mg, 0.30 mmol) was heated neat under nitrogen in an isoxazolone as yellow crystals (113 mg, 82%), m.p.= 219 ºC.
1H-NMR (d6– DMSO+CDCl3)(δppm): 1.11 (t, J=7Hz, 3H), 2.9 (q, J=7Hz, 2H), 6.87 (d, J=8.5Hz, 2H), 7.18 (d, J=8.5Hz, 2H), 7.73 (d, J=9.1Hz,1H), 8.40 (dd, J1 =9.1Hz, J2=2.3Hz, 1H), 8.75 (d, J=2.3Hz, 1H), 10.29 (bs, 1H, NH).
13C-NMR (d6-DMSO+CDCl3)(δppm): 7.9, 30.9, 79.05, 114.63, 119.46, 124.11, 132.46, 135.08, 137.21, 143.79, 153.92, 158.31, 161.38, 165.88.
FT-IR : 3140, 2965, 1773, 1683, 1591, 1531, 1324, 1188, 1114, 1010, 961, 832 cm-1.
MS m/z: 432 (M+, 27%), 430(M+, 30%), 406(74), 404(77), 279(100), 251(20), 184(35), 182(36), 157(29), 155(29), 102(22), 72(23), 44(59).
The following compounds were made by the same procedure
4-propionyl-3-(4-methylphenylamino)-2-(5-nitropyridin-2-yl)isoxazole-5(2H)-one (11b).
Yellow needles (85%), m.p.= 157-159 ºC, after recrystalization from ethanol.
1H-NMR (d6– DMSO+CDCl3)(δppm): 1.12 (t, J=7.05Hz, 3H), 2.35 (s, Me, 3H), 2.98 (q, J=7.05Hz, 2H), 7.04 (d, J=8.5Hz,2H), 7.07(d, J=8.5Hz, 2H), 7.07 (d, J= 9.0Hz, 1H), 8.55 (dd, J1=9.0Hz, J2=2.5Hz, 1H), 8.91 (d, J=2.5Hz, 1H), 10.33 (s, 1H, NH).
13C-NMR (d6– DMSO+CDCl3)(δppm): 7.9, 24.66, 30.9, 81.05, 115.42, 122.40, 134.74, 135.36, 136.83, 141.89, 143.92, 154.28, 160.88, 163.62, 164.19
FT-IR: 3177, 1762, 1700, 1600, 1515, 1338, 1208, 1123, 976, 838 cm-1.
MS m/z: 368.1 (M+, 13%), 354 (100), 294 (57), 269 (16), 248 (40), 230 (16), 220
(16), 158 (39), 144 (13), 118 (21), 117 (20), 107 (16), 91 (67), 78 (16), 65 (20), 44 (33).
4-propionyl-3-(4-methoxyphenylamino)-2-(5-nitropyridin-2-yl)isoxazole-5(2H)-one(11c).
Yellow needles (80%), m.p.= 187-189 ºC.
1H-NMR (d6– DMSO+CDCl3)(δppm): 1.11 (t, J=7Hz, 3H), 2.9 (q, J=7Hz, 2H), 3.77 (s, OMe, 3H), 6.79 (d, J=8.7Hz, 2H), 7.10 (d, J=8.7Hz, 2H), 7.52 ( d, J=9.0Hz,1H), 8.54(dd, J1=9 Hz, J2=2.1Hz, 1H), 8.93 (bd, J=2.1, 1H), 10.26 (s,1H, NH).
13C-NMR (d6– DMSO+CDCl3)(δppm): 7.9, 30.9, 55.89, 84.87, 114.85, 115.59, 130.74, 141.93, 143.95, 154.33, 158.40, 161.38, 163.69, 164.32, 175.2.
FT-IR: 3823, 1785, 1700, 1592, 1345, 1207, 1115, 1030, 838 cm-1.
MS m/z: 384.1 (M+, 10%), 360 (100), 310 (49), 295 (43), 264 (21), 249 (13), 221 (14), 193 (12), 194 (10), 174 (21), 146 (10), 134 (34), 133 (22), 123 (17), 92 (16), 77 (29), 44 (37).
1-(2-(4-bromophenylamino)-6-nitroimidazo[1,2-a] pyridine-3-yl) propan-1-one(13a).
The isoxazolone 9a (99 mg, 0.23 mmol) and triethylamine (0.3 mL) were refluxed in ethanol (11 mL) for 1 h. The mixture was left to cool to room temperature and the desired compound was collected as pale cream needles (74.76 mg, 84 %), m.p.= 197 ºC.
1H-NMR (d6– DMSO+CDCl3)(δppm): 1.18 (t, J=7Hz, 3H), 2.44 (q, J=7Hz, 2H), 7.49 (d, J=8.5Hz, 2H), 7.68 (d, J=9.7Hz, 1H), 7.74 (d, J=8.5Hz , 2H), 8.19 (dd, J1=9.7Hz, J2=1.6Hz, 1H), 8.87 (bd, J=1.6, 1H), 9.88 (s,1H, NH).
13C-NMR (d6– DMSO+CDCl3)(δppm): 7.6, 32.8, 99.72, 114.62, 114.97, 121.76, 123.93, 132.38, 137.66, 140.01, 147.17, 155.28, 160.87.
FT-IR: 3285, 2955, 1643, 1611, 1555, 1475, 1331, 1294, 1201, 1102, 1079, 1002, 820 cm-1.
MS m/z : 388 (M+, 62%), 386 (M+, 64%), 360 (5), 358 (6), 279 (100), 251 (16), 233 (14), 205 (12), 184 (11), 182 (12), 157 (12), 155 (12), 102 (14), 78 (13), 77 (11).
1-(2-(4-methylphenylamino)-6-nitroimidazo[1,2-a] pyridine-3-yl) propan-1-one (13b).
The above procedure using 9b (85 mg, 0.26 mmol) and triethylamine (0.3 mL) gave the desired imidazole as pale cream needles (66.4 mg, 75%), m.p.=188-189 ºC.
1H-NMR (d6– DMSO+CDCl3)(δppm): 1.18 (t, J=7Hz, 3H), 2.35 (s, 3H, Me), 2.44 (q, J=7Hz, 2H), 7.19 (d, J=8.2Hz, 2H, Ar), 7.51 (d, J=9.7Hz, 1H, Ar), 7.59 (bd, J=8.2Hz, 2H, Ar), 8.15 (bd, J=6.7Hz, 1H, Ar), 8.85 (bs, 1H, NH), 9.84 (bs, 1H, Ar).
13C-NMR (d6– DMSO+CDCl3)(δppm): 7.6, 24.3, 32.8, 98.99, 114.30, 119.33, 122.80, 127.26, 130.07, 132.93, 137.11, 147.34, 157.76, 161,15.
FT-IR: 3455, 1662, 1608, 1555, 1308, 1208, 1015, 822 cm-1.
MS m/z: 324.1 (M+, 100%), 317(48), 248(27), 220(9), 144(10), 118(13), 91(20), 78(6),
65(6).
1-(2-(4-methoxyphenylamino)-6-nitroimidazo[1,2-a]pyridine-3-yl) propan-1-one (13c) and
1-( 5- methoxy-2-(4-nitrophenylamino)indol-3-yl) propan-1-one (14).
The isoxazolone 9c (96 mg, 0.25 mmol) and triethylamine (0.3 mL) were refluxed in ethanol (11 mL) for 3 h. On cooling to room temperature, the precipitate was filtered to give an orange solid(72.20 mg), shown (NMR and TLC) to be a mixture of two compounds, which were separated by silicagel p.l.c eluting three times with dichloromethane. The first band was separated and washed with n-hexane to give 12 as orange needles (18.80 mg, 20%), m.p.= 211-214 ºC.
1H-NMR (d6– DMSO+CDCl3)(δppm): 1.18 (t, J=7Hz, 3H), 2.44 (q, J=7Hz, 2H), 3.72 (s, OMe, 3H), 6.86 (dd, J1=8.7Hz,J2=2.5Hz,1H), 6.92(d, J=9.1Hz, 1H), 7.32 (d, J=8.7Hz, 1H), 7.45 (bs, 1H), 8.42 (dd, J1=9.1Hz, J2=2.6Hz,1H), 9.26 (d, J=2.6Hz, 1H), 10.72 (bs, 1H, NH).
13C-NMR (d6– DMSO+CDCl3)(δppm): 7.6, 32.8, 55.13, 103.86, 111.00, 111.69, 112.14, 125.67, 126.89, 133.76, 145.26, 145.59, 156.41, 156.58.
FT-IR: 3345, 1642, 1615, 1500, 1331, 1215, 1117, 1035, 838 cm-1.
MS m/z: 340.1 (M+, 70%), 324 (100), 295 (27), 264 (32), 249 (13), 221 (29), 193 (12), 194 (10), 150 (13), 78 (6), 77 (5), 40 (5). The second band was separated and washed with n-hexane to give 11c as a red solid (53.4 mg, 59%), m.p.= 161-162 ºC.
1H-NMR (d6– DMSO+CDCl3)(δppm): 1.18 (t, J=7Hz, 3H), 2.44 (q, J=7Hz, 2H), 3.72 (s, OMe, 3H), 6.95 (d, J=8.9Hz, 2H), 7.50 (d, J=9.7Hz, 1H) 7.62 (bd, J=8.9Hz, 2H), 8.16 (dd, J1=9.7Hz, J2=1.9Hz, 1H), 8.78 (bs, 1H, NH), 10.1 (bs, 1H, NH), 9.88 (bs, 1H).
13C-NMR (d6– DMSO+CDCl3)(δppm): 7.6, 34.6, 55.97, 98.76, 114.19, 114.82, 121.31, 122.86, 127.24, 132.85, 137.15, 147.48, 156.16, 160.86.
FT-IR: 3130, 1685, 1615, 1515, 1315, 1222, 1199, 1092, 824 cm-1.
MS m/z: 339.1 (M+, 100%), 315 (34), 295 (43), 264 (12), 249 (12), 221 (10), 194 (8), 193 (11), 134 (16), 92 (8), 90 (8), 78 (8).
Acknowledgements
We thanks Islamic Azad University branch of Piranshahr for financial support.
References
- Khalafy, J.; Prager, R. H. Some unusual reactions of 3-phenylaminoisoxazolones, J. Sci. I. R. Iran, 2000, 11, 32-38.
- Khalafy, J.; Prager, R.H.; Smith, J.A. Photochemical and thermal reactions of isoxazolones substituted at C-3 or C-4 with nitrogen, oxygen or sulfur, J. Chem. Res. (M), 1999, 518-536.
- Doleschall, G. A new enamine-salt for the synthesis of ?-alkylated ethyl acetoacetates, Tetrahedron Lett. , 1988, 29, 6339-6340.
- Azimi, Ch; Sepehraddin, F; Tahazadeh, H; Oriental Journal of Chemistry, 2013, 29, 1443.
- Worral, D.E. The reaction of hydroxylamine and hydrazine on the aryl monothioamides of carbethoxyethylmalonate, J. Am. Chem. Soc. 1923, 45, 3092-3094.
- Barnikow, G; Kunzek, H. Sodium and nickel salts of N-aryl methanecarboxylic acid diethyl ester thioamides, Justus Liebigs Ann. Chem., 1966, 700, 36-45.
- Walter, W.; Meyer, H.W.; Lehmann, A. Thermodynamic acidity of thioanilides, Justus Liebigs Ann. Chem., 1974, 765-775.
- Tateja, N.; Pham, T.; Tuteja, R.; Ocham, A.; Falaschi, A. Inhibition of DNA unwinding and ATPase activities of human helicase II by chemotherapeutic agents, Biochem. Biophys. Res. Commun. 1997, 236, 636-640.
- Stiborova, M.; Bieler, C.A.; Wiessler, M.; Frei, E. The anticancer agent ellipticine on activation by cytochrome P450 forms covalent DNA adducts, Biochem. Pharmacol., 2001, 62, 1675-1684.
- Perrin, D.D. and Amarego, W.L.F. Purification of Laboratory Chemicals, Pergamon Press: Oxford, U.K.; 3rd edn, 1988.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


