An Efficient and Telescopic Process for Saxagliptin Hydrochloride.
1Innovation Plaza, IPDO, R&D, Dr. Reddy’s Laboratories Ltd., Survey Nos.42, 45,46 &54, Bachupally, Qutubullapur, R. R. Dist.-500073, Andhra Pradesh., India
2Department of Chemistry, Osmania University, Hyderabad- 500 007, Andhra Pradesh, India
3Research and development, Macleods Pharmaceuticals Ltd, G-2, Mahakalicaves Road, Andheri East, Mumbai-400093, Maharastra, India
DOI : http://dx.doi.org/10.13005/ojc/300137
Download this article as:
![]()
An efficient and telescopic process for the preparation of saxagliptin hydrochloride (1) is described using T3P as a reagent for condensation and dehydration steps.
KEYWORDS:Saxagliptin; dipeptidyl peptidase-4 inhibitor; propylphosphonic anhydride (T3P)
INTRODUCTION
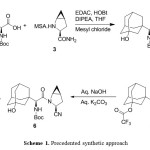
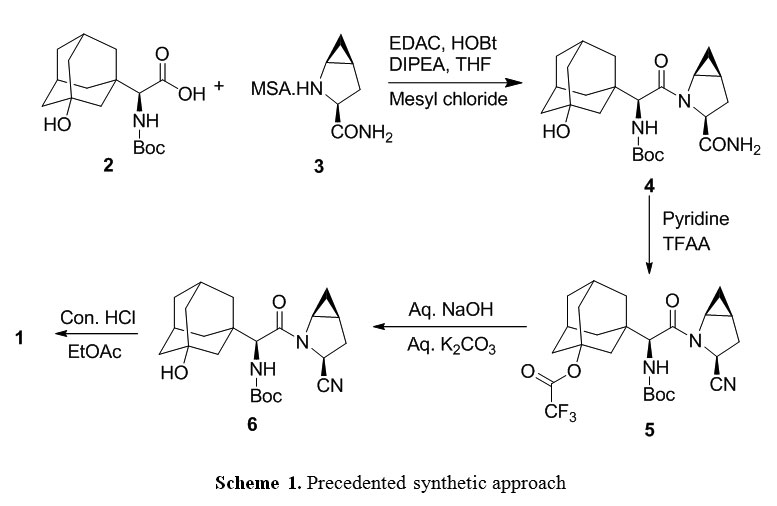
Dipeptidyl peptidase-4 (DPP4) is an enzyme, responsible for the inactivation of the incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulin tropic polypeptide (GIP). Dipeptidyl peptidase 4 (DPP-4) inhibitors enhance the body’s own ability to control blood glucose by increasing the active levels of incretin hormones in the body. DPP-4 inhibitors are clinically attractive for numerous reasons, including a low risk of hypoglycemia when used as monotherapy because of their glucose-dependent mechanism of action, relatively low risk for drug-drug interactions, weight neutrality, simple daily oral administration, lack of a need for dose titration, and a minimal need for therapeutic monitoring. Saxagliptin 1 is a dipeptidyl peptidase-4 inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus and it is approved by United States of Food and Drug Administration (USFDA) in the form of 2.5 and 5 mg base film-coated tablets under the brand name of Onglyza®. The reported2 synthetic route for synthesis of 1 as shown in scheme 1, where (S)-2-((tert-butoxycarbonyl).
  |
Scheme 1. Precedented synthetic approach Click here to View Scheme |
amino)-2 ((1r,3R,5R,7S)-3-hydroxyadamantan-1-yl)acetic acid compound (2) was condensed with (1S,3S,5S)-2-azabicyclo[3.1.0]hexane-3-carboxamide methanesulfonate compound (3) using 1-Ethyl-3-(3-dimethyllaminopropyl) carbodiimide hydrochloride. (EDC.HCl) as coupling agent in presence of 1-hydroxybenzotriazole (HOBt) furnished (tert-butyl ((S)-2-((1S,3S,5S)-3-carbamoyl-2-azabicyclo[3.1.0]hexan-2-yl)-1-((1r,3R,5R,7S)-3-hydroxyadamantan-1-yl)-2-oxoethyl)carbamate (4). Thereafter, compound 4 was subjected to dehydration using trifluoroacetic anhydride (TFAA) and pyridine afforded compound (1R,3r,5R,7S)-3-((S)-1-((tert-butoxycarbonyl)amino)-2-((1S,3S,5S)-3-cyano-2-azabicyclo[3.1.0]hexan-2-yl)-2-oxoethyl)adamantan-1-yl 2,2,2-trifluoroacetate (5), which upon treated with aq. NaOH and aq. K2CO3 obtained tert-butyl ((S)-2-((1S,3S,5S)-3-cyano-2-azabicyclo [3.1.0]hexan-2-yl)-1-((1r,3R,5R,7S)-3-hydroxyadamantan-1-yl)-2-oxoethyl)carbamate (6).
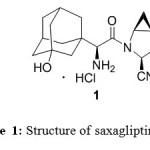 |
Figure 1: Structure of saxagliptin HCl Click here to View Figure |
Finally, compound 6 was treated with hydrochloric acid to afford 1. In addition the first reported3 procedure an alternative process for dehydration of 4 using imidazole & phosphorous oxy chloride (POCl3) in presence of pyridine was reported. Despite the proven potential of reported procedures for the synthesis of 1, there are certain disadvantages associated with the reported procedures including (i) usage of hazardous chemicals such as TFAA, POCl3, and pyridine. (ii) Use of numerous reagents, which are needed purifications or acid base treatments for removal such as HOBt, TFAA. (iii) requirement of stringent water content for the moisture sensitive reagents such as EDC.HCL, (iv) multiple steps and (v) purifications including column chromatography for the removal of impurities generated during the reaction or carried from the raw materials and for the reagents that are used in the reaction. We have developed a process using propylphosphonic anhydride (T3P)4 as a reagent for condensation and dehydration steps and finally Boc deprotection of 6 and simultaneous HCl salt formation leads to saxagliptin hydrochloride.
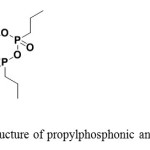 |
Figure 2: Structure of propylphosphonic anhydride (T3P) Click here to View Figure |
RESULTS AND DISCUSSION
At our end to develop robust process for the preparation of 1, we started with condensation step as shown in scheme 2 to avoid the drawbacks associated with the condensation step in reported procedures as mentioned above and various coupling agents were screened such as cyanuric chloride5, N,N’-carbonyldiimidazole (CDI)6, 2-Chloro-4,6-dimethoxy-1,3,5-triazine (CDMT)7, diethyl chlorophosphate (DECP)8, 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM)9, Methanesulfonyl chloride (MsCl) & HOBt2, EDC.
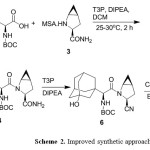 |
Scheme 2. Improved synthetic approach Click here to View Scheme |
HCl-HOBT and propylphosphonic anhydride (T3P)10. Experimental results revealed that T3P and EDC-HOBt offer better yield and purity when compared with other coupling reagents as shown in Table 1.
Table1: Condensation of 2 and 3 with different coupling agents
|
Entry |
Reagent |
Base |
Solvent |
Reagent equiv. |
cPurity (%) |
|
1 |
Cyanuric chloride |
aNMM |
EtOAc |
0.6 |
22.78 |
|
2 |
CDI |
bDIPEA |
DCM |
1.1 |
20.18 |
|
3 |
CDMT |
NMM |
EtOAc |
1.0 |
58.68 |
|
4 |
DECP |
DIPEA |
EtOAc: CAN |
1.0 |
— |
|
5 |
DMT-MM |
NMM |
MeOH : DCM |
1.1 |
79.14 |
|
6 |
MsCl/HOBT |
DIPEA |
THF |
1.1/0.05 |
— |
|
7 |
EDC/HOBT |
DIPEA |
EtOAc |
1.16/1.07 |
93.40 |
|
8 |
T3P |
DIPEA |
DCM |
1.0 |
94.55 |
T3P is commercially available non-toxic, non-allergenic coupling agent used for amide bond linkage, it offers broad functional-group tolerance and low epimerization tendency and salt by-products that are formed during the reaction are non-hazardous, that are completely water soluble. Additionally, usage of multiple reagents and laborious work up procedures such as acid base treatments and purifications for the removal of by products can be avoided by replacing reported coupling agents with T3P. It is commercially available with 50% of concentration 11 in different solvents systems such as DCM, THF, EtoAc, DMF etc. The obtained compound 4 was confirmed by spectral data (Mass & NMR). Solvent always plays pivotal role on yield and quality of the end product. In view of this, to check the impact of T3P reagent in various solvents including polar aprotic solvents (DMF), ether solvents (THF), acetate solvents (EtOAc) and chlorinated solvents (DCM) were examined. Optimal results were obtained in DCM when compared with other solvents as depicted in Table 2
Table 2: Condensation of 2 and 3 with different T3P solutions
|
Entry |
aReagent |
Solvent |
Temp (°C) |
Equiv |
bPurity (%) |
|
1 |
T3P in DMF |
DMF |
40-45 |
1 |
86.95 |
|
2 |
T3P in THF |
THF |
40-45 |
1 |
90.96 |
|
3 |
T3P in EtOAc |
EtOAc |
40-45 |
1 |
93.41 |
|
4 |
T3P in EtOAc |
EtOAc |
25-35 |
1 |
92.79 |
|
5 |
T3P in DCM |
DCM |
40-45 |
1 |
93.42 |
|
6 |
T3P in DCM |
DCM |
25-35 |
1 |
94.55 |
Having developed an efficient process for 4, our attention was turned towards the dehydration of 4. The reported processes involved the usage of TFAA & pyridine, combination of imidazole, POCl3 & pyridine as dehydrating agents for the conversion of 4 to 6. Dehydration of 4 by using TFAA as dehydrating agent proceed through the ester compound 5 as an intermediate, which is an additional step and hydrolysis of ester compound 5 needed a strong base. To avoid the usage of a noxious carcinogen base (pyridine) and highly toxic reagents such as POCl3 and TFAA and to circumvent the disadvantageous associated with the reporting procedures, screened the various dehydrating reagents such as cyanuric chloride12, triethylammonium sulphate (TEA.SO3)13, DECP14, 2-Chloro-4,6-dimethoxy-1,3,5-triazine (CDMT (Chloromethylene)dimethy liminium chloride (Vilsmeier Reagent)15 and T3P. Experimental results revealed that T3P and TFAA offer better yield and purity when compared with the other reagents as shown in Table 3. Safety, handling and process efficiency point of view T3P found to be better reagent when compared to TFAA.
Table 3: Dehydration of 4 with different regents
|
Entry |
Reagent |
Solvent |
Temp (°C) |
Yield (%) |
Time (h) |
aPurity (%) |
|
1 |
Cyanuric chloride |
DMF |
25-30 |
— |
48 |
— |
|
2 |
TEA.SO3 |
TEA |
25-30 |
— |
24 |
— |
|
3 |
DECP |
Toluene |
25-30 |
— |
24 |
— |
|
4 |
CDMT |
DMF |
70-75 |
— |
24 |
— |
|
5 |
Vilsmeier reagent yliminium chloride |
ACN:DMF |
25-30 |
— |
10 |
— |
|
6 |
T3P |
DCM |
25-30 |
83 |
7 |
85.71 |
|
7 |
TFAA |
EtOAc |
25-30 |
80 |
1 |
61.89 |
To further improve the efficiency of process, as per our strategy different solvents including DMF, THF, EtOAc, and DCM were examined. DCM as a solvent furnished the product in highest yield and purity as shown in Table 4 entries 5 and 6.
Table 4: Dehydration of 4 with T3P in different solvents
|
Entry |
aReagent |
Solvent |
Temp(°C) |
Time (h) |
Equiv |
bPurity (%) |
Yield (%) |
|
1 |
T3P in DMF |
DMF |
70-75 |
7 |
2 |
— |
— |
|
2 |
T3P in THF |
THF |
60-65 |
8 |
2 |
— |
— |
|
3 |
T3P in EtOAc |
EtOAc |
75-80 |
8 |
2 |
70.16 |
78 |
|
4 |
T3P in EtOAc |
EtOAc |
25-35 |
48 |
2 |
50.01 |
71 |
|
5 |
T3P in DCM |
DCM |
40-45 |
7 |
2 |
88.22 |
82 |
|
6 |
T3P in DCM |
DCM |
25-35 |
24 |
2 |
94.36 |
83 |
This modification was eliminated the laborious work up process and allowed us to develop an one pot synthesis for the penultimate intermediate 6 in single solvent system without isolation of amide compound of 4. Absence of acid proton at δ 12.4 of 2 and amine proton at δ 2.5 of 3 indicating the peptide bond formation, further absence of amide protons of 3 at δ 4.6 and presence of peak at δ 119.3 in 13C NMR of 6 indicating the cyano carbon and confirms the formation of 6. The obtained compound 6 was further substantiated by 1H-1H correlation (gDQ-COSY, Figure 3) and 1H-13C correlation (gHSQC, Figure 4). Telescoped the process by the direct conversion of 2 and 3 to 6 using T3P and DIPEA alone it self for both the stages and observed incompletion of reaction during dehydration step and performed the reaction at elevated temperature in order to completion of the reaction and ended up with impurities formation. Finally the obtained compound 6 from the above process was subjected to Boc deprotection with aq. HCl followed by HCl salt formation as per the literature process2 leads to saxagliptin HCl. Absence of Boc protons at δ 1.14 in 1H NMR and δ 155.8 in 13C NMR indicating the formation of 1 and the obtained results are congruence with reported values of 1.
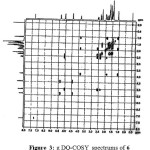 |
Figure 3: g DQ-COSY spectrums of 6 Click here to View Figure |
CONCLUSIONS
We have developed simple, cost effective, robust, high yielding and safer process for the synthesis of saxagliptin hydrochloride 1 that has following advantages over the reported processes; (i) usage of hazardous reagents such as trifluoroacetic anhydride (TFAA), phosphorous oxy chloride (POCl3), pyridine etc. are replaced with non-toxic, non-allergenic and commercially available propylphosphonic anhydride (T3P) (ii) usage of numerous reagents, which are needed purifications or acid base treatments for removal such as HOBt, TFAA are avoided (iii) column chromatography purifications for the removal of the related compounds/impurities are avoided and (iv) one pot synthesis was developed for the penultimate intermediate of saxagliptin hydrochloride 1.
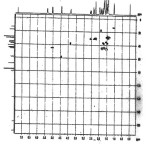 |
Figure 4: g HSQC spectrums of 6 Click here to View Figure |
EXPERIMENTAL SECTION
Solvents and reagents were obtained from commercial sources and used without further purification. The 1H and 13C spectra were measured in DMSO-d6, CD3OD and CDCl3 using 400 and 100 MHz on a Varian Gemini and Varian Mercury plus 2000 FT NMR spectrometer, g-DQ-COSY and gHSQC experiments were performed to identify the 1H-1H correlation and 1H-13C correlation, the chemical shifts were reported in δ ppm. IR spectra were recorded in the solid state as KBr dispersion using Perkin-Elmer 1650 FT IR spectrometer. The mass spectrum (70 eV) was recorded on an HP 5989 A LC-MS spectrometer. The solvents and reagents were used without further purification. Given the current socioeconomic and environmental concerns, commercial synthetic approach to an active pharmaceutical ingredient (API) needs to be safer, greener, simple, cost effective and less energy intensive. Therefore, in order to develop business driven commercial route as early as possible in the lifecycle of a compound, where even modest cost improvements can result in large savings in the cost of manufacturing a drug substance, a meticulous scientific approach is required.
Tert-butyl ((S)-2-((1S,3S,5S)-3-cyano-2-azabicyclo [3.1.0]hexan-2-yl)-1-((1r,3R,5R,7S)-3-hydroxyadamantan-1-yl)-2-oxoethyl)carbamate (6)
To a stirred solution of 3 (20 g, 0.089 mol) in DCM (200 mL) was added 2 (29.3 g, 0.089 mol) and DIPEA (62.6 mL 0.359 mol) at 25-30°C and maintained for 10-15 min. To the resultant mixture, T3P in DCM 50% solution (28.6 g .089 mol) was added at 25-30°C over a period of 15 min and maintained for 2h. After completion of the reaction added water (200 mL) and stirred for 10-15 min. Separated organic layer and washed the organic layer with water (100 mL) followed by 5% aq. NaHCO3 (100 mL) solution. The organic layer thus obtained was concentrated under vacuum at 25-35°C to afforded 4 in the form of crude. Dissolved the compound 4 in DCM (200 mL) and added DIPEA (62.6 mL, 0.036 mol) followed by T3P in DCM 50% solution ( 2 x 28.6 g, 0.089 mol ) in two lots with the time interval of 7 h at 25-30oC and maintained the reaction at same temperature for 24 h. Monitor the reaction by TLC and after completion of the reaction added water (300 mL) and stirred for 10-15 min. Separated organic layer and washed the organic layer with water (150 mL) followed by 5% aq. NaHCO3 solution (300 mL). Distilled off organic layer under vacuum at 25-35°C afforded 6 in the form of off white coloured solid. Purity by HPLC: 94.36%; yield: 32.4 g (83 %); MS (m/z): 416.2 (M+1)+; FT-IR (KBr) Cm−1: 3569, 3544 (O-H stretching), 3380 (N-H stretching), 2245 (CN stretching), 1704, 1650 (C=O stretching), 1248 (C-N stretching), 1167 (C-O stretching); 1H NMR (400 MHz, CDCl3):δ 1.07 (m, 2H, CH2), 1.43 (s, 9H, CH3), 1.48-1.80 (m, 12H, CH2), 1.89 (m, 1H, CH), 2.25 (br, 2H, CH), 2.37-2.58 (m, 2H, CH2), 3.86 (m, 1H, CH), 4.46 (d, 1H, J=10, CH), 5.04 (dd, 1H, J=2.0, 10.4), 5.38 (d, 1H, J=10, NH); 13C NMR (100 MHz, CDCl3) : δ 13.5, 17.8, 28.4, 30.2, 30.5, 35.2, 37.1, 37.5, 38.0, 41.2, 44.3, 44.4, 45.1, 46.2, 58.6, 68.5, 80.0, 119.3, 155.8, 170.0.
Saxagliptin hydrochloride.
To a stirred solution of 6 (25 g, 0.060 mol) in ethyl acetate (150 mL) was added aq. HCl (11N,130 mL) at 25-30 °C and maintained at same temperature for 4 h. Separated solid was filtered, washed with ethyl acetate (50 mL) and dried at 40 °C under vacuum for 5 h to afford 1. Yield: 19.6 g (92.3%). Purity by HPLC: 99.85%, MS (m/z): 316.4 (M+1)+; FT-IR (KBr) Cm−1: 3448, 3374 (O-H stretching), 2747, 2671, 2610, 2094 (N+-H stretching), 1642 (C=O stretching), 2242 (CN stretching), 1242 (C-N stretching), 1033 (C-O stretching); 1H NMR (DMSO-d6, 400 MHz, ) :δ 0.96-1.13 (m, 2H, CH2), 1.62-1.77 (m, 12H, CH2), 2.01 (m, 1H, CH), 2.28 (br, 2H, CH), 2.33-2.65 (m, 2H, CH2), 3.97 (td, J=2.4, 6.0, CH), 4.30 (s, 1H, CH), 5.20 (dd, 1H, J=2.4 Hz, 10.8 Hz, CH); 13C NMR (100 MHz, CD3OD) :δ 14.9, 19.8, 31.9, 32.0, 36.4, 38.6, 39.9, 41.4, 45.2, 45.3, 47.1, 47.5, 60.5, 69.2, 121.0, 167.9.
ACKNOWLEDGMENTS
We thank the management of Dr. Reddy’s Laboratories Ltd. for supporting this work. Co-operation from the colleagues of analytical research & development department and process research & development department of active pharmaceutical ingredients, IPDO, Dr. Reddy’s Laboratories Ltd. is highly appreciated.
REFERENCES
- Idris I, Donnelly R. Diabetes, Obes. Meta., 9, 153-65 (2007). von Geldern T. W, Trevillyan J. M, Drug Dev. Res. 67, 627-642(2006).
- Scott A.S, Gregory S.J, Sergei K, Shelly A.R, Truc V, Robert E.W. Org. Process Res. Dev. 13(6), 1169-76(2009).
- David J.A, Jeffrey A.R, David A.B, David R.M, Ashish K, James G.R, Aiying W, Ligaya M.S, Prakash T, Qi H, Song – Ping H, Benoni A.O, Michael C, Li X, Li T, Effie T, Gustav E.W, Donald M.E, Jovita M, Shu Y.C, Scott A.B, Mark S.K, Rex A.P, Lawrence G.H. J. Med. Chem. 48, 5025-37(2005).
- Wissmann H, Kleiner H. J. Angew. Chem., Int. Ed. Engl. 19, 133(1980). Boggs S.D, Cobb J.D, Gudmundsson K.S, Jones L.J, Matsuoka R.T, Millar A, Patterson D.E, Samano V, Trone M.D, Xie S, Zhou X.-M. Org. Process Res. Dev. 11,539-45 (2007).
- Grzegorz B. Tetrahedron 62, 9507 (2006).
- Paul R, Anderson G.W. J. Am. Chem. Soc., 82, , 4596 (1960). Emily K.W, Julian G.K.C, Philips A.H, David R.J.H, John P.G. Org. Process Res. Dev.13, 106-13 (2009).
- Heather L.R, Lisa F.Org. Process Res. Dev. 3, , 172-76 (1999). Kaminski Z.J. Tetrahedron Lett. 26, 2901(1985). Kaminski Z.J. Synthesis 917 (1987). Taylor E.C, Schrader T.H, Walensky L.D. Tetrahedron 48,19 (1992).
- Won J.E, Kim H.K, Kim J.J, Yim H.S, Kim M.J, Kang S.B, Chung H.A, Lee S.;G, Yoon Y.J. Tetrahedron. 63, 12720 (2007).
- Kunishima M.K, Iwasaki C.F.T, Tani K.S. Tetrahedron Lett. 40, 5327 (1999). Kunishima M.K, Hioki C.K, Terao K, Tani S. Tetrahedron. 57, 551 (2001). Wen-Chung S, Zhuoliang C, Song X, Joe M, Run-Ming W, Kapa P, Oljan R. Tetrahedron Lett. 49, 5359 (2008).
- Helal, C. J.; Kang, Z.; Lucas, J. C.; Bohall, B. R. Org. Lett. 6, 1853 (2004).
- Christopher M, Melanie B, Christine S, Jens H. T3P: e-EROS Encyclopedia of Reagents for Organic Synthesis Propylphosphonic Anhydride, (2012)
- Aquino F, Pauling H, Walther W, Plattner DA, Bonrath W. Synthesis 5, 731 (2000). Fabrice A, Reinhard K, Horst P, Werner B. Molecules. 2, 176 (1997).
- Kazunori W, Atsushi N, Akira I, Yoshinori N, Nobuji N, Shoji F, Yoo T. Tetrahedron 59, 5337 (2003).







ISSN Online: 2231-5039

![]()




{kind=link}