Geometrical Study of a New Cobalt(III) Complex, fac-[Co(HEAC)2 ]ClO4 , Containing Eight Chiral Centers with Mixed Donor (NN’O) Ligand
Mohammad Hakimi
Chemistry Department, Payame Noor University, 19395-4697 Tehran, I. R. Iran.
In this work, a new cobalt(III) complex, [Co(HEAC)2 ]ClO4 ’”2.5H2O (1), with the HEAC, 2- (2-(2-hydroxyethylamino)ethylamino)cyclohexanol, was prepared and identified by elemental analysis, FT-IR and UV-Vis spectroscopy, cyclic voltammetry and single-crystal X-Ray diffraction. The cobalt atom in the crystal structure of 1 has a distorted octahedral geometry by coordination of the four nitrogen and two oxygen atoms of HEAC. The cyclic voltammetry of 1 revealed two reversible oxidation and reduction steps, corresponding to the CoIII/CoII and CoII/Co0 couple. The complex 1 has one center of inversion on the cobalt atom and Ci symmetry. The axial Co”N bond lengths are similar to equatorial one, proving low spin status for d6 configuration and no Jahn-Teller effects. Possible six diastereomers in complexation of HEAC to cobalt atom are present in this paper.
KEYWORDS:Cobalt(III) Complex; Amino Alcohol; Cyclic Voltammetry; Ci Symmetry; X-ray Crystal Structure
Download this article as:| Copy the following to cite this article: Hakimi M. Geometrical Study of a New Cobalt(III) Complex, fac-[Co(HEAC)2 ]ClO4 , Containing Eight Chiral Centers with Mixed Donor (NN’O) Ligand. Orient J Chem 2013;29(1). |
| Copy the following to cite this URL: Hakimi M. Geometrical Study of a New Cobalt(III) Complex, fac-[Co(HEAC)2 ]ClO4 , Containing Eight Chiral Centers with Mixed Donor (NN’O) Ligand. Available from: http://www.orientjchem.org/?p=11944 |
Introduction
The synthesis of β-amino alcohols is of current interest as they are important sub-units commonly used in the synthesis of biologically active natural and synthetic products, pharmaceuticals, synthetic amino acids and pesticides1−2 and as cross-linking reagents in the synthesis of precursor systems for superconductors3.
In the past few years we have studied complexation of multi N-donor ligands4−7. In continuation of our previous studies, in this work the preparation of cobalt(III) complex, [Co(HEAC)2]ClO4∙2.5H2O (1), with 2-(2-(2-hydroxyethylamino)ethylamino)cyclohexanol (HEAC, scheme 1) was described. This complex was characterized by elemental analysis, FT-IR and UV-Vis spectroscopy, cyclic voltammetry and X-ray crystallography.
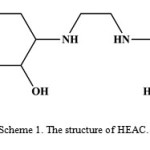 |
Scheme 1. The structure of HEAC. |
Material and Methods
General methods
All starting chemicals and solvents were reagent or analytical grade and used as received. The HEAC was prepared accordingly to the literature8. A conventional three–electrode system voltammetry was employed, with a PGSTAT101 and glassy carbon working electrode. The infrared spectrum of a KBr pellet was recorded in the range 400–4000 cm−1 using a FT-IR 8400-Shimadzu spectrometer. The carbon, hydrogen and nitrogen contents were determined in a Thermo Finnigan Flash Elemental Analyzer 1112 EA. Melting point was determined using a Barnsted Electrothermal 9200 electrically heated apparatus. The electronic spectrum was recorded in H2O using a Shimadzu model 2550 UV-Vis spectrophotometer (190–900 nm).
Synthesis of [Co(L)2]ClO4∙2.5H2O, 1.
A solution of HEAC (2 mmol, 0.40 g) in methanol (25 mL) was mixed with CoCl2∙6H2O (1 mmol, 0.24 g), to produce a purple solution. O2 was bubbled through the solution for 12 h, then the solution was left at room temperature for 2 d. Then it was diluted to l.5 L with water, and sorbed onto a column of Sephadex-SP C-25 cation exchange resin. Elution of a fast-moving major purple band was achieved with 0.2 M NaClO4.Crystals suitable for X-ray diffraction were grown from a dilute water solution. Yield (0.51 g) 84 %; m.p.: 148 °C. Anal. Calcd for C20H47ClCoN4O10.5(%): C, 39.64; H, 7.82; N, 9.25. Found: C, 40.11; H, 7.77; N, 9.42. IR (KBr, cm−1): 3267 s (ν OH), 3069 m (ν NH), 2928 s (ν CH), 2827 m (ν CH2), 1445 m (δas CH2), 1350 m (δs CH2), 1230 w (ν CO), 1121 s (ν1 ClO4), 1116 s (ν CN), 1025 (ν4 ClO4), 988 (ν2 ClO4), 815 (ν3 ClO4) and 626 (ν5 ClO4) cm−1. UV-Vis (H2O, λmax (nm)/ε): 229/6727 (n→π*), 401/99 (1A1g→1T2g), 553/81 (1A1g→1T1g). Cyclic voltammetry (glassy carbon, 100 mV s−1 scan rate): E½ (CoIII/CoII) −0.32 V, E½ (CoII/Co0) −1.19 V vs. Ag/AgCl.
Crystal structure determination and refinement
The data collection for 1 was carried with a Bruker APEX-II CCD diffractometer, using graphite-monochromatized MoKα (λ = 0.71073 Å) radiation at 296 K. The data were integrated with SAINT9 and corrected for Lorentz and polarization effects. Absorption was corrected for using SADABS10. The structure was solved by Patterson methods, implemented in SHELXS-9711. Refinement by full-matrix least-squares methods based on F2 values against all reflections has been performed by SHELXL-9711, including anisotropic displacement parameters for all non-H atoms. The position of hydrogen atoms belonging to the carbon atoms Csp2 were geometrically optimized applying the riding model [Csp2–H, 0.93 Å; Uiso(H) = 1.2 Ueq(C)]. Calculations concerning the molecular geometry, the verification of space group and the analysis of hydrogen bonds were performed with PLATON12. Table 1 contains crystallographic data and details of the data collection and structure refinement. Selected bond lengths (Å) and angles (°) are listed in table 2.
Table 1. Crystal data and structure refinement for 1.
| Empirical formula | C20H47ClCoN4O10.5 |
| Formula weight (g mol−1) | 606.00 |
| Temperature (K) | 293 |
| Crystal system | Triclinic |
| Space group | 12P1″> |
| Unit cell dimensions (Å, °) | |
| a | 9.5363(6) |
| b | 9.7054(6) |
| c | 17.6096(8) |
| α | 85.119(4) |
| β | 87.865(4) |
| γ | 63.567(6) |
| Volume (Å3), Z | 1454.15(14), 2 |
| Calculated density (g cm−3) | 1.384 |
| Absorption coefficient (mm−1) | 0.74 |
| F(000) | 646 |
| Crystal size (mm3) | 0.22 × 0.16 × 0.04 |
| θ range for data collection (°) | 2.7–29.1 |
| h, k, l ranges | −12:12, −12:9, −22:23 |
| Reflections collected | 11969 |
| Independent reflections | 6512 |
| Rint | 0.053 |
| Data / restraints / parameters | 6512 / 99 / 380 |
| Goodness-of-fit on F2 | 0.90 |
| Final R indices [I > 2σ(I)] | R1 = 0.079, wR2 = 0.1989 |
| R indices (all data) | R1 = 0.1848, wR2 = 0.229 |
| Largest diff. peak and hole (e.Å−3) | 0.83 and −0.40 |
Table 2. Selected bond length (Å) and angles (°) for 1 with estimated standard deviations in parentheses.
| Distances | Angles | ||
| Co1A–O1A | 1.897(3) | O1A–Co1A–N1A | 93.2(2) |
| Co1A–N1A | 1.959(4) | O1A–Co1A–N2A | 96.0(2) |
| Co1A–N2A | 1.979(5) | N2A–Co1A–N1A | 85.8(2) |
| Cl1–O1 | 1.359(9) | O1–Cl1–O3 | 108.3(6) |
| Cl1–O2 | 1.383(6) | O3–Cl1–O2 | 107.1(5) |
| Cl1–O3 | 1.39(1) | O2–Cl1–O4 | 108.0(5) |
| Cl1–O4 | 1.419(6) | O4–Cl1–O1 | 109.1(5) |
Results and Discussion
The reactions between methanolic solution of HEAC and cobalt(II) chloride in presence of O2 provided purple crystals of 1. It is air-stable and soluble in DMSO and H2O.
The ν (N–H) in the IR spectrum of 1 shifted 28 cm−1 to lower energy respect to the free ligand, indicating coordination through the nitrogen atoms. The characteristic frequencies corresponding to the perchlorate group13 appear at about 1121 (ν1), 1025 (ν4), 988 (ν2), 815 (ν3) and 626 (ν5) cm−1.
UV–Vis spectrum of 1 in aqueous solution exhibited a broad absorption with the λmax at 229 nm for n→π* transition, 401 and 553 nm for d→d transitions of the cobalt(III) complex.
Cyclic voltammetry data for the cobalt(III) complex in the range of −0.32 to −1.19 V and 0.10 M TBAP-DMSO solution are listed in the experimental section. The cyclic voltammogram of 1 showed two reversible oxidation and reduction waves, corresponding to the CoIII/CoII and CoII/Co0 couple.
Description of the crystal structure
The crystal structure of 1 was determined by X-ray single-crystal diffraction. The molecular graphics were drawn with ORTEP-III14 and Diamond15.
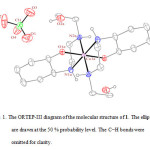 |
Figure 1: The ORTEP-III diagram of the molecular structure of 1. The ellipsoids are drawn at the 50 % probability level. The C−H bonds were omitted for clarity. |
In the crystal structure of 1 (figure 1), the cobalt atom has a distorted octahedral coordination environment. Four sites are occupied by nitrogen atoms with the Co–N bond lengths in the range of 1.959(4)–1.979(5) Å. The two other sites are occupied by two oxygen atoms with the Co–O bond lengths 1.897(3) Å. The hydroxyl group of the cyclohexyl ring is coordinated to the metal ion while the other is clearly not. The complex 1 has one center of inversion which is placed on the cobalt atom and Ci symmetry (figure 2).
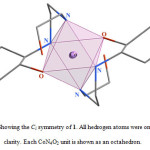 |
Figure 2. Showing the Ci symmetry of 1. All hedrogen atoms were omitted for clarity. Each CoN4O2 unit is shown as an octahedron. |
The cobalt(III) ion has d6 configuration in low spin status and no Jahn-Teller effects. The axial Co−N bond lengths are similar to equatorial ones, proving this issue. Of particular interest from coordination chemistry perspective asymmetrical HEAC ligand, is that several diastereomers are possible in complexation. There are six possible arrangement of the pair of ligands around the octahedral metal ion exist (scheme 2) for octahedral coordination of two HEAC ligand as tridentate chelate. Among these possible diastereomers, the all-trans–fac form was observed in 1.
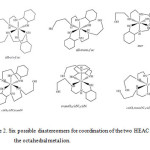 |
Scheme 2. Six possible diastereomers for coordination of the two HEAC around the octahedral metal ion. |
Supplementary material
CCDC 748469 for [Co(L)2]ClO4∙2.5H2O (1) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html.
Acknowledgement
We are grateful to Payame Noor University of I. R. Iran for financial support.
References
- Ager D. J., Prakash I. and Schaad D. R., Chem. Rev., 96, 835 (1996).
- Corey E. J. and Zhang F.-Y., Angew. Chem., Int. Ed., 38, 1931 (1999).
- Wang S., Trepanier S. J., Zheng J. C., Pang Z. and Wagner M. J., Inorg. Chem., 31, 2118 (1992).
- Hakimi M., Mardani Z., Moeini K., Minoura M. and Raissi H., Z. Naturforsch., 66b, 1122 (2011).
- Hakimi M., Mardani Z., Moeini K., Mohr F., Schuh E. and Vahedi H., Z. Naturforsch., 67b, 452 (2012).
- Hakimi M., Maeder M. and Lawrance G. A., J. Coord. Chem., 64, 105 (2011).
- Hakimi M., Kukovec B.-M., Normohammadzadeh Z., Schuh E. and Mohr F., J. Chem. Crystallogr., 42, 180 (2012).
- Hakimi M., Mardani Z., Moeini K. and Fernandes M. A., J. Coord. Chem., 65, 2221 (2012).
- SAINT (version 7.12), Bruker Analytical X-ray Instruments Inc., Madison, Wisconsin, USA (2004).
- Sheldrick G. M., SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Gottingen, Gottingen, Germany (1996).
- Sheldrick G. M., Acta Crystallogr., A46, 467 (1990).
- Spek A. L., J. Appl. Crystallogr., D65, 148 (2009).
- Hakimi M., Moeini K., Mardani Z., Fernandes M. A., Mohr F. and Schuh E., J. Coord. Chem. 65, 1232 (2012).
- Farrugia L. J., J. Appl. Crystallogr., 30, 565 (1997).
- Bergerhoff G., Berndt M. and Brandenburg K., J. Res. Natl. Inst. Stand. Technol., 101, 221 (1996).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


