Ft-Ir, Ft-Raman and Quantum Chemical Calculations of 1-Phenylpyrrole
R. Renjith1, J. B. Bhagysree3, Rajeev. T. Ulahannan1, Hema Tresa Varghese2 and C. Yohannan Panicker1
1Department of Physics, TKM College of Arts and Science, Kollam, India. 2Department of Physics, Fatima Mata National College, Kollam, India. 3Department of Chemistry, Mar Ivanios College, Nalanchira, Trivandrum, Kerala, India.
The IR and Raman spectra of the title compound have been recorded and analyzed. The harmonic vibrational wavenumbers were calculated theoretically using Gaussian09 software package. Calculations were performed by HF and DFT levels using the standard 6-31G* basis sets. The data obtained from vibrational wavenumber calculations are used to assign vibrational bands found in the IR and Raman spectra of the title compound. The calculated wavenumbers (DFT) agree well with the observed wavenumbers. The predicted infrared intensities, Raman activities and first hyperpolarizability are reported.
KEYWORDS:DFT; pyrrole; hyperpolarizability
Download this article as:| Copy the following to cite this article: Renjith R, Bhagysree J. B, Ulahannan R. T, Varghese H. T, Panicker C. Y. Ft-Ir, Ft-Raman and Quantum Chemical Calculations of 1-Phenylpyrrole. Orient J Chem 2013;29(1). |
| Copy the following to cite this URL: Renjith R, Bhagysree J. B, Ulahannan R. T, Varghese H. T, Panicker C. Y. Ft-Ir, Ft-Raman and Quantum Chemical Calculations of 1-Phenylpyrrole. Orient J Chem 2013;29(1). Available from: http://www.orientjchem.org/?p=25208 |
Introduction
Substituted pyrroles represent an important class of five membered heterocycles that are the structural moieties found in several natural products and in therapeutically active compounds including fungicides1-5, antibiotics6-8, cholesterol reducing agents9 and antitumor drugs10. The pyrrole subunit also plays an important role in material science as the building block of polypyrroles, an important representative class of conducting polymers, which have found wide applications in the area of new materials due to their chemical, thermal and electrical properties associated to their easiness and low cost of production11. Multisubstituted 1-phenylpyrrole derivatives are among the building blocks of active pharmaceutical ingredients having cytostatic12,13, antiviral14, antibiotic activities or mineral corticoid receptor antagonist effect15. Faigl et al.16 reported the combined application or regioselective brominations and organometallic reactions to prepare several multisubstituted derivates of phenylpyrroles. Santos and Silva17 reported a calorimetric and computational study of the thermochemistry of halogenated 1-phenylpyrrole derivatives. In the present study the FT-IR, FT-Raman and theoretical calculations of the wavenumbers of the title compound are reported.
Experimental
The FT-IR spectrum was recorded using a DR/Jasco FT-IR 6300 spectrometer. The spectral resolution was 2 cm-1. The FT-Raman spectrum was obtained on a Bruker RFS 100/s, Germany. For excitation of the spectrum the emission of Nd:YAG laser was used, excitation wavelength 1064 nm, maximal power 150 mW.
Computational Details
Calculations of the title compound were carried out with Gaussian09 software program18 using the HF/6-31G* and B3LYP/6-31G* basis sets to predict the molecular structure and vibrational wavenumbers. The DFT hybrid B3LYP functional method tends to overestimate the fundamental modes; therefore scaling factors have to be used for obtaining a considerably better agreement with experimental data19. The wavenumber values computed contain known systematic errors and we therefore, have used the scaling factor values of 0.8929 and 0.9613 for HF and DFT basis sets19. The assignment of the calculated wavenumbers is aided by the animation option of Gaussview program, which gives a visual presentation of the vibrational modes20.
Results and Discussion
IR and Raman spectra
The observed IR, Raman bands and assignments are given in table 1. Phenyl CH stretching modes21 are expected above 3000 cm-1 and for the title compound, the bands observed at 3146, 3109 cm-1 in the Raman spectrum are assigned as CH stretching modes of the phenyl ring. The DFT calculations give these modes at 3188, 3181, 3157, 3144, 3108 cm-1. The benzene ring possesses six ring stretching vibrations, of which the four with the highest wavenumbers (occurring near 1600, 1580, 1490 and 1440 cm-1) are good group vibrations. With heavy substituents, the bands tend to shift to somewhat lower wavenumbers. In the absence of ring conjugation, the band at 1580 cm-1 is usually weaker than that at 1600 cm-1. In the case of C=O substitution, the band near 1490 cm-1 can be very weak. The fifth ring stretching vibration is active near 1315 ± 65 cm-1, a region that overlaps strongly with that of the CH in-plane deformation. The sixth ring stretching vibration, or the ring breathing mode, appears as a weak band near 1000 cm-1, in mono-, 1,3-di- and 1,3,5-trisubstituted benzenes. In the otherwise substituted benzenes, however, this vibration is substituent sensitive and difficult to distinguish from the ring in-plane deformation21,22. The υPh modes are expected in the region 1285-1610 cm-1 for mono substituted benzenes21,22. The DFT calculations give the Ph stretching modes at 1600, 1583, 1461, 1458, 1318 cm-1. The υPh modes are observed at 1605, 1466, 1455, 1299 (IR) and at 1597, 1468 cm-1 (Raman) for the phenyl ring. The ring-breathing mode of monosubstituted benzenes21 appears near 1000 cm-1, and the band at 1011 cm-1 theoretically is assigned to this mode.
The out-of-plane CH deformation bands of the phenyl ring are assigned at 988, 960, 911, 828, 726 cm-1 theoretically. According to literature, the out-of-plane CH deformation near 770 ± 50 cm-1 and the out-of-plane ring deformation near 690 cm-1 form a pair of strong bands characteristic of mono substituted benzene derivatives21. In the present case, these bands are observed at 722 and 683 cm-1 in IR spectrum. The in-plane CH deformation modes of the benzene ring are observed at 1261, 1188, 1133, 1022 cm-1 in the IR spectrum, 1195, 1060, 1030 cm-1 in the Raman spectrum and at 1290, 1188, 1176, 1067, 1026 cm-1 theoretically (DFT).
The CN stretching vibrations23 are observed in the region 1330-1260 cm-1 due to stretching of the phenyl carbon nitrogen bond and in the range 950-1100 cm-1. The CN stretching mode is reported at 1192 cm-1 experimentally and at 1228, 1219 and 1195 cm-1 as theoretically24. In the present case, the C3-N12 stretching band is observed at 1236 cm-1 in the IR spectrum, 1256 cm-1 in the Raman spectrum and at 1244 cm-1 theoretically. The other CN stretching bands are assigned at 1081 and 1074 cm-1 theoretically. For the title compound the CC stretching modes are observed at 922 cm-1 in the IR spectrum and at 993, 921 cm-1 theoretically as expected23.
The pyrrole CH stretching modes are observed at 3098, 3077, 3055 cm-1 in the IR spectrum, 3073 cm-1 in the Raman spectrum and at 3102, 3095, 3081, 3073 cm-1 theoretically21,23. The in-plane and out-of-plane CH modes are also identified and assigned (Table 2). The substituent sensitive modes are also identified and assigned (table 1).
First hyperpolarizability
Non-linear optics deals with the interaction of applied electromagnetic fields in various materials to generate new electromagnetic fields, altered in wavenumber, phase or other physical properties25. Many organic molecules, containing conjugated π electrons and characterized by large values of molecular first hyperpolarizabilities, were analyzed by means of vibrational spectroscopy26,27. Analysis of organic molecules having conjugated π-electron systems and large hyperpolarizability using infrared and Raman spectroscopies has evolved as a subject of research 28 .First hyperpolarizability is a third rank tensor that can be described by a 3 3 3 matrix. The 27 components of the 3D matrix can be reduced to 10 components due to the Kleinman symmetry29. The calculated first hyperpolarizability of the title compound is 4.98 10-30 esu.. We conclude that the title compound is an attractive object for future studies of non linear optical properties.
In order to investigate the performance of vibrational wavenumbers of the title compound, the root mean square (RMS) value between the calculated and observed wavenumbers were calculated. The RMS values of wavenumbers were calculated using the following expression30.
The RMS= 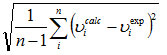 error of the observed IR and Raman bands are found to 31.68, 37.60 for HF and 11.37, 6.37 for DFT methods, respectively. The small differences between experimental and calculated vibrational modes are observed. This is due to the fact that experimental results belong to solid phase and theoretical calculations belong to gaseous phase.
error of the observed IR and Raman bands are found to 31.68, 37.60 for HF and 11.37, 6.37 for DFT methods, respectively. The small differences between experimental and calculated vibrational modes are observed. This is due to the fact that experimental results belong to solid phase and theoretical calculations belong to gaseous phase.
Frontier molecular orbitals
The analysis of the wavefunction indicates that the electron absorption corresponds to a transition from the ground to the first excited state and is mainly described by one electron excitation from the HOMO to LUMO. Both the HOMO and the LUMO are the main orbital taking part in chemical reaction. The HOMO energy characterizes the capability of electron giving; LUMO characterizes the capability of electron accepting31. The frontier orbital gap helps to characterize the chemical reactivity, optical polarizability and chemical hardness-softness of a molecule32. Surfaces for the frontier orbitals were drawn to understand the bonding scheme of the title compound. The calculated HOMO and LUMO energies are -8.773 and -54.566 eV. The chemical hardness and softness of a molecule is a good indication of the chemical stability of the molecule. From the HOMO-LUMO energy gap, one can find whether the molecule is hard of soft. The molecules having large energy gap are known as hard and molecules having a small energy gap are known as soft molecules. The soft molecules are more polarizable than the hard ones because they need small energy to excitation. The hardness value of a molecule31 can be determined as η = (-HOMO+LUMO)/2. The value of η of the title molecule is 2.104 eV. Hence we conclude that the title compound belongs to hard material.
 |
Figure 1 |
Table 1. Calculated wavnumbers (scaled), observed IR and Raman bands and assignments.
| HF/6-31G* | B3LYP/6-31G* | IRυ(cm-1) | Ramanυ(cm-1) | Assignments | ||||
| υ(cm-1) | IR intensity | Raman activity | υ(cm-1) | IR intensity | Raman activity | |||
| 3105 | 1.96 | 162.49 | 3188 | 1.55 | 151.11 | υCHPh | ||
| 3097 | 2.97 | 0.20 | 3181 | 2.06 | 0.19 | υCHPh | ||
| 3076 | 7.60 | 91.77 | 3157 | 7.60 | 119.78 | υCHPh | ||
| 3063 | 7.31 | 92.42 | 3144 | 5.57 | 98.25 | 3146 | υCHPh | |
| 3034 | 5.42 | 289.46 | 3108 | 4.62 | 292.02 | 3109 | υCHPh | |
| 3028 | 20.47 | 0.28 | 3102 | 18.33 | 0.22 | υCH | ||
| 3020 | 33.35 | 55.59 | 3095 | 31.37 | 66.35 | 3098 | υCH | |
| 3008 | 9.40 | 118.35 | 3081 | 10.39 | 124.71 | 3077 | υCH | |
| 2999 | 1.43 | 36.86 | 3073 | 1.94 | 38.48 | 3055 | 3073 | υCH |
| 1617 | 70.90 | 90.92 | 1600 | 61.57 | 158.27 | 1605 | υPh | |
| 1597 | 7.60 | 5.89 | 1583 | 5.43 | 3.493 | 1597 | υPh | |
| 1538 | 1.28 | 1.26 | 1521 | 0.24 | 3.57 | 1512 | υC=C | |
| 1509 | 196.63 | 13.02 | 1501 | 136.60 | 30.27 | υC=C | ||
| 1476 | 63.96 | 20.88 | 1461 | 74.12 | 35.39 | 1466 | 1468 | υPh |
| 1460 | 6.34 | 1.15 | 1458 | 4.16 | 1.74 | 1455 | υPh | |
| 1397 | 3.68 | 35.36 | 1391 | 9.56 | 24.32 | 1399 | 1402 | δCH |
| 1348 | 0.13 | 0.44 | 1343 | 0.10 | 0.54 | 1350 | δCH | |
| 1321 | 112.36 | 91.32 | 1320 | 105.58 | 172.69 | 1322 | 1323 | δCH |
| 1298 | 0.29 | 0.50 | 1318 | 0.00 | 2.66 | 1299 | υPh | |
| 1261 | 2.07 | 1.11 | 1290 | 0.33 | 0.05 | 1261 | δCHPh | |
| 1215 | 0.91 | 0.02 | 1244 | 3.39 | 0.59 | 1236 | 1256 | υCN |
| 1182 | 0.60 | 3.93 | 1188 | 1.97 | 6.62 | 1188 | 1195 | δCHPh |
| 1145 | 0.65 | 8.23 | 1176 | 0.29 | 7.15 | 1133 | δCHPh | |
| 1111 | 20.14 | 11.22 | 1111 | 15.97 | 8.81 | 1099 | 1114 | δCH |
| 1080 | 5.62 | 6.10 | 1081 | 1.65 | 1.98 | 1088 | υCN | |
| 1072 | 8.24 | 0.25 | 1074 | 17.30 | 3.58 | 1077 | υCN | |
| 1053 | 1.46 | 1.08 | 1067 | 29.19 | 15.49 | 1060 | δCHPh | |
| 1049 | 54.80 | 9.78 | 1026 | 11.28 | 1.21 | 1022 | 1030 | δCHPh |
| 1026 | 0.01 | 0.79 | 1011 | 13.50 | 0.44 | 1011 | υPh | |
| 1020 | 2.49 | 0.59 | 993 | 2.72 | 48.77 | υCC | ||
| 1009 | 11.02 | 0.24 | 988 | 0.56 | 0.73 | 987 | γCHPh | |
| 986 | 10.87 | 39.06 | 960 | 0.25 | 1.16 | γCHPh | ||
| 965 | 10.69 | 1.99 | 921 | 18.23 | 23.23 | 922 | υCC | |
| 956 | 0.06 | 1.75 | 911 | 6.96 | 2.47 | 914 | γCHPh | |
| 920 | 20.83 | 20.95 | 880 | 0.06 | 1.27 | γCH | ||
| 918 | 0.21 | 1.60 | 872 | 0.08 | 4.87 | 871 | γCH | |
| 879 | 0.41 | 5.33 | 838 | 0.00 | 9.03 | γCH | ||
| 878 | 0.10 | 5.12 | 828 | 0.93 | 0.03 | 830 | 829 | γCHPh |
| 796 | 55.24 | 0.02 | 766 | 46.44 | 0.28 | 762 | γCH | |
| 771 | 138.22 | 2.32 | 726 | 102.92 | 3.15 | 722 | γCHPh | |
| 770 | 0.88 | 3.17 | 697 | 29.25 | 1.14 | δPh | ||
| 714 | 39.99 | 1.19 | 691 | 1.35 | 4.95 | δRing | ||
| 685 | 5.88 | 3.87 | 686 | 7.54 | 3.47 | 683 | 670 | γPh |
| 646 | 21.75 | 2.78 | 635 | 7.28 | 3.18 | δRing | ||
| 621 | 5.88 | 2.60 | 619 | 3.97 | 1.82 | δPh | ||
| 617 | 1.54 | 0.22 | 608 | 2.10 | 0.79 | 609 | δRing | |
| 540 | 11.00 | 1.39 | 524 | 7.82 | 1.18 | δPh | ||
| 423 | 0.30 | 0.45 | 420 | 0.21 | 1.08 | δPh | ||
| 412 | 0.27 | 1.03 | 414 | 0.43 | 0.99 | δRing | ||
| 347 | 0.27 | 5.54 | 352 | 0.58 | 4.90 | γPh | ||
| 297 | 0.39 | 5.65 | 286 | 0.22 | 5.74 | 278 | δRing | |
| 134 | 0.01 | 4.31 | 141 | 0.02 | 3.79 | tPh | ||
| 101 | 0.61 | 2.22 | 100 | 0.33 | 1.58 | tRing | ||
| 58 | 0.98 | 8.47 | 67 | 1.14 | 8.30 | tRing | ||
Ph-phenyl ring; Ring-pyrrole ring; υ-stretching; δ-in-plane deformation; γ- out-of-plane deformation; t-torsion.
Conclusion
The IR and Raman spectra of the title compound have been recorded and analyzed. The harmonic vibrational wavenumbers were calculated theoretically using Gaussian09 software package. Calculations were performed by HF and DFT levels using the standard 6-31G* basis sets. The calculated wavenumbers (DFT) agree well with the observed wavenumbers. The data obtained from vibrational wavenumber calculations are used to assign vibrational bands found in the IR and Raman spectra of the title compound. The predicted infrared intensities, Raman activities and first hyperpolarizability are reported.
References
- Koyama, M., Ohtani, N., Kai, F., Moriguchi, I., Inouye, S., J. Med. Chem. 30: 552 (1987).
- Ziogas, B.N., Kalmarakis, A.E., J. Phytopathol. 149: 301 (2001).
- Forster, B., Staub, T., Crop Prot. 15: 529(1996).
- Irmler, S., Rogniaux, H., Hess, D., Pillonel, C., Pestic. Biochem. Physiol. 84: 25 (2006).
- Noguchi, R., Banno, S., Ichikawa, R., Fukumori, F., Ichiishi, A., Kimura, M., Yamaguchi, I., Fujimura, M., Fungal Genet. Biol. 44: 208 (2007).
- Kaneda, M., Akamura, S., Ezaki, N., Iitaka, Y., J. Antibiot. 34: 1366 (1981).
- Koyama, M., Kodama, Y., Tsuruoka, T., Ezaki, N., Niwa, T., Inouye, S., J. Antibiot. 34: 1569 (1981).
- Koyama, M., Ezaki, N., Tsuruoka, T., Inouye, S., J. Antibiot. 36: 1483 (1983).
- Roth, B.D., Prog. Med. Chem. 40: 1 (2002).
- Denny, W.A., Rewcastle, G.W., Baguley, B.C., J. Med. Chem. 33: 814 (1990).
- Street, G.B., Skotheim, T.A., Handbook of conducting polymers, vol.1, Marcel Dekker, New York (1986).
- Remers, W.A., Rao, S.N., Singh, U.C., Kollman, A., J. Med. Chem. 29: 1256 (1986).
- Campiani, G., Morelli, E., Fabbrini, M., Nacci, V., Greco, G., Novellino, E., J. Med. Chem. 42: 4462 (1999).
- Genin, M.J., Allwine, D.A., Anderson, D.J., Barbachyn, M.R., Emmert, D.E., Garmon, S.A., Graber, D.R., Grega, K.C., Hester, J.B., Hutchinson, D.K., Morris, J., Reischer, R.J. Ford, C.W., Zuerenko, G.E., Hamel, J.C., Schaadt, R.D., Stapert, D., Yagi, B.H., J. Med. Chem. 43: 953 (2000).
- Tatsuta, K., Itoh, M., J. Antibiot. 47: 602 (1994).
- Faigl, F., Matravolgyi, B., Deak, S.,.Holczbauer, T., Czugler, M., balazs, L., Hermecz, I., Tetrahedron 68: 4259 (2012)
- Santos, A.F.L.O.M., Silva, A.V.R., J. Chem. Thermodynamics, 42: 1441 (2010).
- Frisch, M.J., et al., Gaussian 09, Revision B.01., Gaussian, Inc., Wallingford CT (2010).
- Foresman, J.B., in: Frisch, E.,(Ed.) Exploring Chemistry with Electronic Structure Methods: A Guide to using Gaussian, Pittsburg, PA (1996).
- Dennington, R., Keith, T., Millam, J., Gaussview, Version 5, Semichem Inc. ShawneeMissionKS (2009).
- Roeges, N.P.G., A Guide to the Complete Interpretation of the Infrared Spectra of Organic Structures, Wiley, New York (1994).
- Varsanyi, G., Assignments of vibrational Spectra of Seven Hundred Benzene Derivatives, Wiley, New York (1974).
- Silverstein, R.M., Bassler, G.C., Morril, T.C., Spectrometric identification of organic compounds, ed.5, John Wiley and Sons, Singapore (1991).
- Bhagyasree, J.B., Varghese, H.T., Panicker, C.Y., Samuel, J., Van Alsenoy, C., Bolelli, K., Yildiz, I., Aki, E., Spectrochim. Acta 102: 99 (2013).
- Shen, R., The Principles of Nonlinear Optics, Wiley, New York (1984).
- Kolinsky, P.V., Opt. Eng. 31: 1676 (1992).
- Eaton, D.F., Science 253: 281 (1991).
- Tommasini, M.,.Castiglioni, C, Del Zoppo, M., Zerbi, G., J. Mol. Struct. 480: 179 (1999).
- Kleinman, D.A., Phys. Rev. 126:1977 (1962).
- Joseph, T., Varghese, H.T.,.Panicker, C.Y., Viswanathan, K., Sundaraganesan, N., Subramanina, N., Dolezal, M., Global J. Anal. Chem. 3:1(2012).
- Fukui, K., Science 218: 747 (1982).
- Kosar, B., Albayrak, C., Spectrochim. Acta 78A: 160 (2011)

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


