Effect of Added Dimethyl Sulphoxide on the Solvolysis of Cinnamoyl Chloride in Aqueous Acetone and Acetonitrile
G. Raveendran and S. Neelakumari
Department of Chemistry, Sree Narayana College, Kollam - 691 001, India.
Kinetic studies on solvolysis of cinnamoyl chloride in aqueous acetone and acetonitrile with lesser water content shows an order 2.6 with respect to water and it is comparable to that of benzoyl chloride. The relatively greater enthalpy and lesser negative entropy of activation compared to benzoyl chloride favours an SN2 mechanism for cinnamoyl chloride rather than addition elimination mechanism. A sharp increase in rate observed in presence of small amounts of DMSO is due to its, strong basic and hydrogen bonding character leading to the liberation of strong nucleophile OH-, relatively greater stabilization of transition state than intial state which lowers the enthalpy and free energy of activation, distruction and interpenetration into the solvation shell of water and its direct interaction with the substrate.
KEYWORDS:Cinnamoyl chloride; Addition-Elimination; Solvolysis; Hydrolysisl; SN2; SN1; Enthalpy and Entropy of Activation; Dipolar aprotic solvents; Dimethyl sulphoxide
Download this article as:| Copy the following to cite this article: Raveendran G, Neelakumari S. Effect of Added Dimethyl Sulphoxide on the Solvolysis of Cinnamoyl Chloride in Aqueous Acetone and Acetonitrile. Orient J Chem 2012;28(3). |
| Copy the following to cite this URL: Raveendran G, Neelakumari S. Effect of Added Dimethyl Sulphoxide on the Solvolysis of Cinnamoyl Chloride in Aqueous Acetone and Acetonitrile. Available from: http://www.orientjchem.org/?p=22989 |
Introduction
The solvolytic reactions on carboxylic acid derivatives can be basically classified from mechanistic point of view into SN1 and SN2. Inaddition to these two, the possibility of carbonyl addition-elimination mechanism in solvents of low water content was suggested by Williams and Douglas1 which is also called as tetrahedral mechanism. This is due to the presence of a third reactive centre, the carbonyl oxygen, on which electrophilic reagents can interact preferentially leading to a more complex intermediate2. The tetrahedral mechanism may be formulated as follows
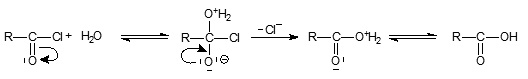
SN1 is occurring in highly polar solvents and in the absence of strong nucleophiles3. SN2 and tetrahedral mechanism may occur in less polar solvents. Whether SN2 or tetrahedral mechanism occurs will depend on the nature of substrate, nucleophile and the leaving group.
The study on hydrolysis of benzoyl chloride and its substituted derivative in aqueous acetone by Berger and Oliver4, Hudson and co-workers.5-10 Zimmerman and Yuan11 states that at relatively low concentrations of water/alcohol in a non-reacting solvent, the reaction was second order with respect to water (or alcohol) and first order with respect to acid halide. The variation of the rates of solvolytic reactions with solvent composition is obtained by plotting the logarithm of the first order rate constant as a function of the logarithm of the molar concentration of the hydroxyl solvent component. The value of the slope is a criterion of mechanism of SN type reactions. The value of the slope is generally 6-7 for SN1 reactions, but much lower, 2-3, for SN2 or addition-elimination reactions. It is seen that the apparent order in water, n, is 2.9 for benzoyl chloride in 2-5 % water- dioxane at 25°C12, 2.72 in
25-40% water – acetone at 25°C, 6.85 in 50-75 % water-acetone and 7.5 in 50-75 % dioxane – water at 0°C. Plot of log k against (D-1)/2 (D+1) (D is the dielectric constant) of the medium is linear up to 30 % water. Above 30 % an increase in slope is observed, which shows a change in mechanism. A change in mechanism is also supported by the observed increase in activation energy with water content. Activation energy gradually increases from 11.73 K.Cal in 5% water to 16.11 K.Cal in 33.3% water. All these observation leads to the conclusion that in solvent rich in acetone (80% aqueous acetone) the hydrolysis of benzoyl chloride proceeds by an SN2 mechanism while in water rich solvent, example in 50% aqueous acetone there is a borderline behaviour in which simultaneous SN1 and SN2 process are occurring and the mechanism can shift to SN1 with further increase in composition of water.
Protic solvents such as fluoro alcohols, hydrogen fluoride, water, methanol, formamide and ammonia are strong hydrogen bond donors while dipolar aprotic solvents, even though are polar, are no more than very weak hydrogen – bond donors. The distinction between protic and dipolar– aprotic solvents is a sharp one. Thus although
N-methylformamide is one of the less protic solvents and nitromethane is one of the less dipolar aprotic, many reactions are more than 100 times faster in nitromethane than in
N-methyl formamide. The classification suggests that hydrogen bonding will be an important interaction in determining protic – dipolar aprotic solvent effects on rates. But other factors, such as dispersion forces, ion – dipole and dipole – dipole interactions, which overlap the protic-dipolar aprotic division, must be taken into account. Many reactions are over a million times faster in dipolar aprotic solvents (DMF) than in protic solvents of much the same dielectric constant. Many dipolar aprotic solvents, eg., DMF, DMSO and HMPT are powerful bases and hydrogen bond acceptors, so that they have strong interactions with solutes which are hydrogen bond donors
According to Lallansingh and his co-workers13 the effect of DMSO on the variation of rate constant with increasing mole percent of the organic co-solvent are due to two factors. They are the solvation changes of the reactants and the transition state. Due to the strong tendency of DMSO to break down the water – water interaction to its aqueous binary mixture, it is quite possible that it may enter into the structure broken region of the solvation sheets of different ionic species. Also DMSO being a dipolar aprotic solvent, it can solvate by ion-dipole type of mechanism, the transition state being large cation, is likely to be solvated more than the initial state. This change in solvation will decrease the activation energy and thereby increase the rate. This accounts for increase of the rate in beginning. The increase of DMSO in the binary solvent mixture decreases the bulk dielectric constant on account of its low value compared to that of water. This may lower the concentration of highly polarized transition state and thereby decrease the rate. This is supported by the values of activation parameters.
According to Murto14, Tommila and Savolainen15 Virtanen16 and Tommila and Pitkanen17 the rate enhancement in DMSO- protic mixtures is due mainly to favourable solvation of transition states by the mixtures relative to pure protic solvent. Tommila and Savolainen have noted that, up to rather high DMSO concentrations, there is sufficient water or alcohol present to form adducts with DMSO and also to completely solvate the hydroxide or methoxide ions. The “rapid” increase in rate of bimolecular reactions involving anion when the molefraction of DMSO in DMSO – H2O mixtures exceeds 0.30 is seen as a result of this. Water forms 2:1 adducts with methanol, 1:1 adducts with DMSO. Roberts18 comes to much the same conclusion, that reactant anion desolvation does not explain the “catalytic” effect of DMSO on ester hydrodysis in aqueous DMSO, rather the effect is due to the ability of DMSO containing solvation shell to stabilize the transition state. Virtanen19 has shown that increasing transition state solvation with increasing mole fraction of DMSO is an important factor in determing the rate of the neutral SN2 hydrolysis of methyl- iodide in DMSO – water mixture.
Experimental
To get a clear picture on the mechanism of hydrolysis of cinnamoyl chloride in aqueous organic solvent and the effect of polar aprotic component like dimethyl sulphxoxide (DMSO) on it, kinetic studies are carried out in aqueous acetone and acetonitrile with percentage of water varying from 5 to 15. To study the effect of aprotic component the solvent choosen is dimethyl sulphoxide whose composition is varied from 0.15M to 0.6M. To determine the activation parameters the rate of the reaction are measured at four different temperatures ranging from 283K to 298K. The cinnamoyl chloride used was Aldrich reagent, and used without distillation or recrystallisation. All solvents used for the kinetics in this work were dried and distilled by the method given in the literature20. Rates were measured conductimetrically
Results and Discussion
Earlier work on benzoyl chloride in aqueous acetone , acetonitrile and dioxane shows that the order with respect to water is 2.7 in aqueous dioxane containing less than 10% water, 2.9 in aqueous acetone containaing less than 40% water and 6.85 in aqueous acetone containing 50-70% water. As already mentioned the value of the slope which can be taken as a criterion for mechanism of SN type reactions is generally 6-7 for SN1 reactions, but much lower, 2-3, for SN2 or addition-elimination reactions. This reveals that in aqueous solution containing lesser amount of water benzoyl chloride undergoes solvolysis by SN2 or addition-elimination mechanism. In the solvolysis of cinnamoyl chloride in aqueous acetone and acetonitrile in the solvent composition 85% to 92.5% of organic component, the order with repect to water is found to be in the range of 2.6(given in the table 1)
Table 1: Determination of order ‘m’ with respect to water for the solvolysis of cinnamoyl chloride in aq. acetone at 283K and 298K.
|
Temperature |
|
Rate const kx104 (sec-1) in |
Value of m | |||
| Solvent | 85% | 87.5% | 90% | 92.5% | ||
|
283K |
Aqueous Acetone |
14.5 |
8.04 |
4.78 |
2.36 |
2.6 |
|
288K |
23.5 |
13.4 |
8.14 |
3.67 |
2.6 |
|
|
293K |
35.6 |
21.4 |
12.5 |
6.41 |
2.6 |
|
|
298K |
62.4 |
35.7 |
19.8 |
10.1 |
2.6 |
|
|
283K |
Aqueous Acetonitrile |
58.0 |
36.2 |
18.8 |
8.56 |
2.7 |
|
288K |
89.6 |
60.3 |
31.3 |
15.3 |
2.6 |
|
|
293K |
138.0 |
89.7 |
46.1 |
23.8 |
2.7 |
|
|
298K |
171.7 |
132.0 |
70.8 |
33.6 |
2.7 |
|
These results shows that in the solvolysis of cinnamoyl chloride in this composition range of the solvent the reaction proceeds either by an SN2 or by an addition-elimination mechanism. The order with respect to water is greater than 2 reveals that one of the water molecules is acting as a nucleophile while the remaining molecules of water acts as a general base to deprotonate H+ from the added nucleophile of the transition state complex and/or involved in stabilization of the transition state by hydrogen bonding.The transition state may either be the loose complex formed in SN2 path way or the tight tetrahedral species formed in addition –elimination pathway.
The activation parameters for benzoyl chloride and cinnamoyl chloride in 92.5% aqueous acetone and acetonitrile are determined and used for comparing the possibility of the two mechanisms.
Table 2 : Determination of activation parameter for the solvolysisof benzoyl and cinnamoyl chloride in 92.5% aqueous acetone and acetonitrile.
|
Substrate |
Solvent |
Temperature |
Rate constant kx103 sec-1 |
Activation |
|||
|
Energy (kj/mole) |
Enthalpy (kj/mole) |
Entropy (j/K, mole) |
Free energy (at 25˚C) (kj/mole) |
||||
|
Cinnamoyl Chloride |
92.5% aq. acetone |
283K |
0.236 |
68.68 |
66.26 |
-80.0 |
90.1 |
|
288K |
0.367 |
||||||
|
293K |
0.628 |
||||||
|
298K |
1.01 |
||||||
|
92.5% aq. acetonitrile |
283K |
0.856 |
63.84 |
61.42 |
-88.61 |
87.82 |
|
|
288K |
1.53 |
||||||
|
293K |
2.38 |
||||||
|
298K |
3.36 |
||||||
|
Benzoyl chloride |
92.5% aq. acetone |
288K |
0.0788 |
49.83 |
47.32 |
-170 |
97.98 |
|
293K |
0.110 |
||||||
|
298K |
0.153 |
||||||
|
303K |
0.210 |
||||||
Solvolysis of benzoyl chloride is charecterised by relatively lower enthalpy of activation, very large negative entropy of activation and relatively greater free energy of activation compared to the respective thermodynamic parameters for cinnamoyl chloride in 92.5% aqueous acetone. But the rate of solvolysis of cinnamoyl chloride is about 4.7 times faster (3.67×10-4 sec-1 at 288K) than that of benzoyl chloride at the same temperature (7.88×10-5 sec-1). This decrease in rate in benzoyl chloride is mainly due to the increase in free energy of activation by about 8 kj/mole. However there is a decrease in enthalpy of activation by about 19 kj/mole and an increase in negative entropy of activation by
90jK-1mole-1. SN2 reaction will be characterized by relatively greater enthalpy of activation and low negative entropy of activation due to the formation of a loose complex as transition state. Addition-elimination reaction will be characterized by relatively lower energy of activation and large negative entropy of activation since the transition state formed will be a tighter tetrahedral complex. From the above observation relating to thermodynamic parameters it can be concluded that solvolysis in benzoyl chloride proceeds through a tight tetrahedral transition state and hence an addition elimination mechanism is favoured in this reaction condition. The decrease in rate and increase in free energy of activation of benzoyl chloride is due to greater contribution of large negative entropy of activation which over takes the decrease in enthalpy of activation. Relatively greater enthalpy of activation and smaller negative entropy of activation suggest that solvolysis in cinnamoyl chloride proceeds mainly by SN2 mechanism than through an addition elimination path way.This may be due to increase in distance of the phenyl group from the reaction center in cinnamoyl chloride by a C═C group which decreases the sterric effect by phenyl group on the reaction center and decreases the extent of conjugation of .>C=O group with the aromatic ring. This is confirmed by the fact that eventhough –COOH group in benzoic acid is meta directing (due to –M effect of the group), -CH=CH-COOH group is mainly ortho, para directing.This decrease in stabilization of the reaction center accounts for the greater reactivity of cinnamoyl chloride compared to benzoyl chloride.
Solvolysis of cinnamoyl chloride in aqueous acetonitrile shows a greater rate than in aqueous acetone with same composition of water (8.56×10-4 sec-1 in 92.5% aqueous acetonitrile and 2.36×10-4 sec-1 in aqueous acetone). This may be accounted in terms of the decrease in the free energy of activation in aqueous acetonitrile(lesser by 2 kj/mole). The main contribution towards the decrease in free energy is the decrease in the enthalpy of activation (from 66.26 kj/mole in aqueous acetone to 61.42 kj/mole in aqueous acetonitrile). However an increase in the negative entropy of activation disfavours the decrease in free energy of activation which accounts for the smaller increase rate. An increase in the negative entropy of activation and decrease in the enthalpy of activation which always favours an addition-elimination pathway which leads to the conclusion that in aqueous acetonitrile there is an increase in the contribution of an addition-elimination path way or a decrease in the contribution of SN2 mechanism compared to the reaction in aqueous acetone.
Reaction of acyl chloride in aqueous DMSO is found to be very fast compared to that in aqueous acetone or acetonitrile. Kinetic studies on the solvolysis of cinnamoyl chloride carried out in 85% to 92.5% aq. acetone and acetonitrile in presence of 0.15M, 0.3M, 0.45M and 0.6M DMSO shows that there is an abnormal increase in rate. The rate constant values and the increase in rate with respect to the rate in the absence of DMSO and the order with respect to DMSO are given in the table 3
Table 3: Comparision of the rate consant and determination of order with respect to DMSO for the solvolysis of cinnamoyl chloride in aqueous acetone in the presence and absenceof DMSO at 298K.
|
Solvent |
[DMSO] |
Rate constant (kx103 sec-1) |
Percentage increase in rate |
Order ‘m’ with respect to DMSO |
|
92.5% aq. acetone |
0.00M |
1.01 |
||
|
0.15M |
3.67 |
263 |
0.62 |
|
|
0.30M |
5.27 |
422 |
||
|
0.45M |
6.73 |
566 |
||
|
0.60M |
8.5 |
742 |
||
|
92.5% aq. acetonitrile |
0.00M |
3.36 |
||
|
0.15M |
6.03 |
79 |
0.56 |
|
|
0.30M |
8.71 |
159 |
||
|
0.45M |
10.7 |
219 |
||
|
0.60M |
13.4 |
299 |
Even though the extent of increase in rate is less in aqueous acetonitrile than aqueous acetone in presence of same concentration of DMSO, the order with respect to DMSO is comparable. The comparability of the order with respect to DMSO reveals that there will not have any considerable mechanistic change with change in solvent. The large increase in rate cannot be due to solvent effect of DMSO, since the amount of DMSO added is very small. Since DMSO can behave as a strong hydrogen bond acceptor and a base, it can penetrate into the water structure thus facilitating deprotonation or ionization of water. This leads to the replacement of water by stronger nucleophile OH− ion in the substitution reaction. Such smaller anions like OH− which are strong hydrogen bond acceptors are about 10−4 to 10−10 times less solvated by polar aprotic component like DMSO than H2O itself. The desolvation of OH− increases its activity and increases the rate of formation of the initial loose complex. Another possibility for the rate increase is the stabilization of transition state by DMSO. It was already suggested that the high increase in rate in presence of DMSO may be due to the ability of DMSO containing solvation shell to stabilize the transition state which is further confirmed by Virtanen21. The variation in the solvation of the initial and transition state in presence of DMSO can be confirmed from the activation parameter values given in the table 4.
Table 4 : Determination activation parameter for the solvolysis of cinnamoyl chloride in 92.5% aqueous acetone and acetonitrile containing 0.3M DMSO.
|
Solvent |
Temperature |
Rate constant kx103sec-1 |
Activation |
|||
|
Energy (kj/mole) |
Enthalpy (kj/mole) |
Entropy |
Free energy (at 25˚C) |
|||
|
92.5% aq. acetone |
283K |
1.88 |
52.94 |
50.53 |
-118.1 |
85.69 |
|
288K |
2.83 |
|||||
|
293K |
4.14 |
|||||
|
298K |
5.84 |
|||||
|
92.5% aq. acetonitrile |
283K |
2.73 |
54.66 |
52.24 |
-109.1 |
84.75 |
|
288K |
3.96 |
|||||
|
293K |
5.95 |
|||||
|
298K |
8.74 |
|||||
Earlier works shows that dimethyl sulphoxide can directly react in stoichiometric composition with acid chlorides and thionyl chloride either in the absence or presence of inert solvents to give α-chlorosulphides22-24. Acetyl chlorides with dimethyl sulphoxides in benzene or methylene chloride mainly gives acetic acid and chloromethylmethyl sulphide. However small amount of two other compounds such as acetoxymethyl methyl sulphides (AMMS) and acetic anhydride are also obtained as byproduct in trace amount depending upon the reaction condition. The reaction is assumed to proceed through two intermediates which are in equilibrium with the two reactants. The formation of acetoxymethyl methylsulphide is accompanied by the formation of HCl which is probably associated with dimethyl sulphoxide which is acting as base in this step. Sulphoxides are basic as is indicated by their ability to form salts with strong acids, their hygroscopic nature and their ability to form strong hydrogen bonds. This is supported by the fact that the concentration ratio, [AMMS/AcOH] increases as the concentration of (CH3)2SO increases. Analysis of the result in terms of first and second order rate expression, the indication is that the reaction is more consistent with a second order kinetics, the first order each in DMSO and acid chloride.
Summary and Conclusion
The aim of this work is to establish a clear picture regarding the mechanism of solvolytic reactions of cinnamoyl chlorides, in aqueous organic solvents like acetone and acetonitrile containing limited amount of water. Even though SN1, SN2 and addition elimination mechanisms are possible in the solvolysis of acid chloride, in presence of lesser amount of water there is no possibility of SN1 mechanism.
The order with respect to water in the solvolysis of cinnamoyl chloride in aqueous acetone and acetonitrile in the composition range 85% to 95% is ≈2.6 which is close to the value obtained for benzoyl chloride (2.7) in the same solvent compositions. This supports either an addion-elimination or SN2 mechanism or a competition between them and never favours an SN1 mechanism. Relaltively greater enthalpy of activation in the range of 61-66 kj/mole compared to 47.32 kj/mole for benzoyl chloride and low negative entropy of activation in the range of -80 to -88 jK-1mole-1 compared to -170 jK-1mole-1 for benzoyl chloride favours an SN2 mechanism for cinnamoyl chloride and an addition-elimination path wayfor benzoyl chloride
Presence of dipolar aprotic solvent molecules like dimethyl sulphoxide (DMSO), shows abnormal increase in rate in aqueous acetone. It can thus be concluded that the sharp increase in rate of hydrolysis in presence of DMSO is due to following factors. 1) Due to strong basic and hydrogen bonding character of DMSO, it can generate OH− from water which behaves as a strong nucleophile than water itself. 2)The polar aprotic solvents like DMSO can not only desolvate smaller nucleophile but also solvate and stabilize the bulky transition state there by decreases the activation parameters like enthalpy, entropy and free energy of activation. 3)When DMSO is added to acetone containing water as cosolvent, in the intial state it enters into the solvation shell of water and when the concentration become high it forms hydrogen bonded complex with water. These interactions decrease the activity of both DMSO and water which accounts for the change in order with respect to water in presence of DMSO 4) DMSO can directly react with substrate forming a series of products.
References
- A Williams and K.T. Douglas, chem. Rev, 75, 628 (1975).
- Dilke, Eley and Sheppard, Trans Faraday soc, 46, 61 (1950).
- Bender and Chen. J. Am. Chem. Soc., 85, 30, (1963).
- G. Berger and S.G.J Oliver, Rec. trav. Chim. 46, 516, 861 (1927).
- Hudson & Wardill, J. Chem. Soc. 1729(1950).
- B.L.Archer and R.F.Hudson, ibid, 3259 (1950).
- D.A.Brown and R.F. Hudson, ibid, 883 (1953).
- C.G. Swain and C.B. Scott, J. Am. Chem.soc; 75, 246 (1953).
- C.G. Swain Am. Chem.soc 70, 1124 (1948).
- C.G. Swain and S.D. Ross, J. Am. Chem.soc. 63, 638 (1946).
- G. Zimmerman and C. Yuan, J. Chem. soc., 77, 332 (1955).
- H. Boehme and W. Schuelhoff, Ber, 84, 28 (1951).
- Lallan singh, R.T. Singh and R.C. Jha, J. Indian Chem. Soc., LVII, 1089, (1980).
- J. Murto and A.M. Hiiro, Suomen Kemistilehil B39, 40, (1966).
- E. Tommila and M. Savolainen, Acta. Chem. Scand., 20, 946 (1966).
- P.O. I. Virtanen, Suomen Kemistileht, B 40, 163 (1967).
- E. Tommilla and I.P. Pitkanen, Acta Chem. Sc and 20, 90, (1966).
- C.A. Kingsbury, J. Org. Chem., 29, 3262, (1964).
- D.D. Roberts J. Org. Chem., 31, 4037, (1966).
- J.F. Cotzem ‘Progress in Physical Chemistry’, Vol. 4, A Streitweiser and R.W. Taft, Ed. Interscience, New York, P-57 (1967).
- P.O. Virtnen, Suomen Kemistilehti, B40, 163 (1967).
- Michael Cocivera, Vincenzo Malatesta, Kyu W.Woo and Alden Effio, J.Org. Chem., 43, 1140-1145, (1977).
- Sergio Thea & Giorgio Cevasco, J. Org. Chem. ,53, 4121 – 4122, (1988).
- P.G.Bordwell & Burnet M Pitt, 579, (1955).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


