Synthesis of Novel Symmetric Schiff Bases Using KSF
Nosrat O. Mahmoodi1, Mohammad A. Zanjanchi, Alireza Aliakbar, Tahere Behzadi and Fateme Ghanbari
1Department of Chemistry, Faculty of Sciences, University of Guilan, P. O. Box 1914, Rasht Iran.
Different novel symmetric Schiff bases were synthesized in a simple and environmentally benign method from the reaction of produced Schiff base with various aldehydes and ketones using montmorollonite KSF clay as heterogeneous solid acidic catalyst in good yields.
KEYWORDS:KSF; Hydrazine hydrate; bis Schiff base; terephtalaldehyde
Download this article as:| Copy the following to cite this article: Mahmoodi N. O, Zanjanchi M. A, Aliakbar A, Behzadi T, Ghanbari F. Synthesis of Novel Symmetric Schiff Bases Using KSF. Orient J Chem 2011;27(2). |
| Copy the following to cite this URL: Mahmoodi N. O, Zanjanchi M. A, Aliakbar A, Behzadi T, Ghanbari F. Synthesis of Novel Symmetric Schiff Bases Using KSF. Orient J Chem 2011;27(2). Available from: http://www.orientjchem.org/?p=24864 |
Introduction
Schiff bases represent a class of important compounds in medicinal and pharmaceutical field. They have biological activities such as antibacterial (1), anticancer (2), antifungal (3), and herbicidal activities (4). Schiff bases have a chelating structure and are in demand because they are straightforward to prepare and are moderate electron donors with easily-tunable electronic and steric effects thus being versatile (5).
Montmorillonite clays have been used as catalysts for a number of organic reactions and offer several advantages over classical acids: strong acidity, noncorrosive properties, recyclability, cheapness, mild reaction conditions, high yields and selectivity, and ease of setup and work up (6). Recently, we used montmorillonite KSF for synthesis of pyridazinones and phthalazinones (7-9).
Experimental
Melting points were measured on a Mettler Fp5 apparatus and are uncorrected. The IR spectra were recorded on a Shimadzu IR-470 spectrophotometer. The ¹H NMR and 13C NMR spectra were recorded on a Bruker 500 MHz instrument using solvent as internal standard (CDCl3 at 7.31 ppm and DMSO-d6 at 2.50, 3.21 ppm). The UV-Vis spectra were recorded on Shimadzu UV-2100.
General procedure for the formation of compound 1: A solution of hydrazine hydrate (0.5 g, 10 mmol) in ethanol 5 mL was added dropwise to a stirred solution of terephtalaldehyde (0.67 g, 5 mmol) in ethanol 5 mL at room temperature. A yellow precipitate starts to form immediately upon addition; the stirring was continued for 1 hour. The resulting solid was filtered and washed with EtOH to yield the desired compound.
1,4-bis(hydrazonomethyl)benzene 1: Yield 90%, m. p. 168 0C, yellow solid. IR (KBr, ν/cm-1): 3350, 3200, 3050, 2900, 1630, 1590, 830. 1H NMR (500 MHz, CDCl3) δ: 5.60 (s, 4H), 7.57 (s, 4H), 7.77 (s, 2H). 13C NMR (500 MHz, CDCl3) δ: 111.39, 126.79, 142.96. UV-Vis (EtOH, λmax/nm) 334.5.
General procedure for the formation of compound 2a-3j: Compound 1 (0.16 g, 1 mmol) and related aldehydes or ketones (2 mmol) was added in ethanol 10 ml. This mixture was heated to reflux in the presence of KSF (20 mg) for required time (4-8 hours). During the reaction a precipitate forms which is insoluble in the reaction solvent. After completion of the reaction, the mixture was allowed to cool to room temperature, then filtered, washed with EtOH and dried; recrystallization from DMF afforded the pure product.
1,4-bis(((4-nitrobenzylidene)hydrazono)methyl)benzene 2a: Yield 70%, m. p. > 310 0C, yellow solid. IR (KBr, ν/cm-1): 3100, 2920, 1620, 1590, 1520, 1340, 830. 1H NMR (CDCl3, 500 MHz) δ: 7.72 (d, 4H, J = 8.9 Hz), 8.02 (s, 4H), 8.25 (d, 4H, J = 8.84), 8.71 (s, 2H), 8.76 (s, 2H). Anal. Calcd for C22H16N6O4; C, 61.68; H, 3.76; N, 19.62; O, 14.94; found; C, 61.72; H, 3.74; N, 19.58; O, 14.95. UV-Vis (EtOH, λmax/nm) 347.5.
1,4-bis(((3-nitrobenzylidene)hydrazono)methyl)benzene 2b: Yield 73%, m. p. 230-236 0C, orange solid. IR (KBr, ν/cm-1): 3100, 2920, 1620, 1520, 1350, 830, 730, 670. 1H NMR (CDCl3, 500 MHz) δ: 7.71 (m, 2H), 8.02 (s, 4H), 8.22 (d, 2H, J = 7.4 Hz), 8.38 (d, 2H, J = 7.11), 8.77 (d, 6H, J = 7.51 Hz). Anal. Calcd for C22H16N6O4; C, 61.68; H, 3.76; N, 19.62; O, 14.94; found C, 61.65; H, 3.90; N, 19.65; O, 14.91. UV-Vis (EtOH, λmax/nm) 348.5.
1,4-bis(((2-hydroxybenzylidene)hydrazono)methyl)benzene 2c: Yield 65%, m. p. 298-300 0C, yellow solid. IR (KBr, ν/cm-1): 3350, 2900, 1620, 1590, 1450, 1210, 830. 1H NMR (CDCl3, 500 MHz) δ: 7.02 (t, 2H, J = 6.8 Hz), 7.08 (d, 2H, J = 7.5 Hz), 7.43 (m, 4H), 7.99 (s, 4H), 8.71 (s, 2H), 8.76 (s, 2H), 8.86 (s, 2H). Anal. Calcd for C22H18N4O2; C, 71.34; H, 4.90; N, 15.13; O, 8.64; found C, 71.30; H, 4.94; N, 15.11; O, 8.66. UV-Vis (EtOH, λmax/nm) 375.2.
1,4-bis(((3-hydroxybenzylidene)hydrazono)methyl)benzene 2d: Yield 69%, m. p. decomposed at > 300 0C, orange solid. IR (KBr, ν/cm-1): 3380, 2950, 1620, 1590, 1450, 1210, 830, 780, 680. 1H NMR (DMSO-d6, 500 MHz) δ: 6.94 (s, 2H), 7.33 (s, 2H), 8 (s, 4H), 8.62 (s, 2H), 8.72 (s, 2H), 8.81 (s, 2H), 9.72 (s, 2H). Anal. Calcd for; C22H18N4O2; C, 71.34; H, 4.90; N, 15.13; O, 8.64; found C, 71.31; H, 4.93; N, 15.11; O, 8.67. UV-Vis (EtOH, λmax/nm) 359, 323.5.
1,4-bis(((4-methoxybenzylidene)hydrazono)methyl)benzene 2e: Yield 64%, m. p. 210-214 0C, yellow solid. IR (KBr, ν/cm-1): 2900, 2850, 1620, 1590, 1550, 1500, 1460, 1440, 1250, 1020, 830. 1H NMR (CDCl3, 500 MHz) δ: 3.9 (s, 6H), 7.49 (d, 4H, J = 5.8 Hz), 8 (m, 8H), 8.51 (s, 2H), 8.76 (s, 2H). Anal. Calcd for C24H22N4O2 C, 72.34; H, 5.57; N, 14.06; O, 8.03; found C, 72.31, H, 5.61, N, 14.09, O, 8.01. UV-Vis (EtOH, λmax/nm) 369.2, 358.8.
1,4-bis(((2-hydroxy,3-methoxybenzylidene)hydrazono)methyl)benzene 2f: Yield 70%, m. p. 272-275 0C, orange solid. IR (KBr, ν/cm-1): 3400, 2900, 1620, 1560, 1460, 1260, 1270, 830, 740. 1H NMR (CDCl3, 500 MHz) δ: 3.98 (s, 6H), 6.95 (t, 2H, J = 7 Hz), 7.04 (d, 4H, J = 6.39 Hz), 7.99 (s, 4H), 8.68 (s, 2H), 8.75 (s, 2H), 8.84 (s, 2H). Anal. Calcd for C24H22N4O4 C, 66.97; H, 5.15; N, 13.02; O, 14.87; found C, 66.99; H, 5.12; N, 13.05; O, 14.89. UV-Vis (EtOH, λmax/nm) 363.5.
1,4-bis(((1-phenylethylidene)hydrazono)methyl)benzene 3g: Yield 55%, m. p. 258-262 0C, yellow solid. IR (KBr, ν/cm-1): 3050, 2920, 1620, 1560, 1440, 1360, 830, 760, 680. 1H NMR (CDCl3, 500 MHz) δ: 3.92 (s, 6H), 7.02 (m. 6H), 7.86 (d, 4H, J = 7.1 Hz), 7.95 (s, 4H), 8.73 (s, 2H). Anal. Calcd for C24H22N4 C, 78.66; H, 6.05; N, 15.29; found C, 78.69; H, 6.07; N, 15.26. UV-Vis (EtOH, λmax/nm) 359.
1,4-bis(((1-(4-methoxyphenyl)ethylidene)hydrazono)methyl)benzene 3h: Yield 56%, m. p. > 300 0C, orange solid. IR (KBr, ν/cm-1): 2920, 1620, 1540, 1500, 1250, 1030, 830. 1H NMR (CDCl3, 500 MHz) δ: 2.58 (s, 6H), 3.85 (s, 6H), 6.99 (d, 4H, J = 8.1 Hz), 7.94 (s, 4H), 8.04 (s, 4H), 8.7 (s, 2H). Anal. Calcd for C26H26N4O2 C, 73.22; H, 6.14; N, 13.14; O, 7.50; found C, 73.19; H, 6.16; N, 13.17; O, 7.48. UV-Vis (EtOH, λmax/nm) 373.5.
1,4-bis(((1-(4-nitrophenyl)ethylidene)hydrazono)methyl)benzene 3i: Yield 62%, m. p. 238-240 0C, yellow solid. IR (KBr, ν/cm-1): 2900, 1620, 1590, 1510, 1340, 850, 830. 1H NMR (CDCl3, 500 MHz) δ: 2.54 (s, 6H), 7.94 (s, 4H), 8.09 (d, 4H, J = 8.8 Hz), 8.27 (d, 4H, J = 8.8 Hz), 8.71 (s, 2H). Anal. Calcd for C24H20N6O4 C, 63.15; H, 4.42; N, 18.41; O, 14.02; found C, 63.17; H, 4.40; N, 18.43; O, 14.05. UV-Vis (EtOH, λmax/nm) 357.5.
1,4-bis(((1-(4-hydroxyphenyl)ethylidene)hydrazono)methyl)benzene 3j: Yield 55%, m. p. decomposed at > 240 0C, orange solid. IR (KBr, ν/cm-1): 3400, 2900, 1620, 1560, 1440, 1210, 830. 1H NMR (CDCl3, 500 MHz) δ: 2.4 (s, 6H), 6.83 (d, 4H, J = 9 Hz), 7.79 (d, 4H, J = 8.6 Hz), 7.94 (s, 4H), 8.66 (s, 2H). Anal. Calcd for C24H22N4O2 C, 72.34; H, 5.57; N, 14.06; O, 8.03; found C, 72.31; H, 5.59; N, 14.08; O, 8.01. UV-Vis (EtOH, λmax/nm) 357.5.
General procedure for the formation of complex 4: Cu (OAc)2 (1mmol) was added to a hot solution of the ligand 2c (1mmol) in ethanol (10 ml) containing a few drops of TEA and the resulting mixture was refluxed for 3h. A pale green precipitate separated, which was collected by filtration and washed with ethanol. Yield 55%, m. p., pale green solid. IR (KBr, ν/cm-1): 1610, 1530, 1460, 1440, 1320, 1200, 1140, 960, 900. 820,740. Anal. Calcd for C44H32Cu2N8O4 C, 61.17; H, 3.73; Cu, 14.71; N, 12.97; O, 7.41; found C, 61.14; H, 3.74; Cu, 14.73; N, 12.6; O, 7.2. UV-Vis (EtOH, λmax/nm) 284, 398.5.
Results and Discussion
In contribution, to this Schiff bases synthesis, we first synthesized the one pot Schiff base 1 by reaction of terephtalaldehyde with hydrazine hydrate (Scheme 1) the reaction was completed in the short time and in good yield.
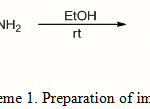 |
Scheme 1: Preparation of imine 1 |
The next step was synthesis of the final Schiff bases by reaction of synthesized amine 1 utilizing different aldehydes and ketones (Scheme 2). In this reaction aldehydes react faster than ketones and the nature of the substituents on the aromatic ring of the aldehydes and ketones has different influences on the reaction rate. For instance, in the case of electron withdrawing groups such as nitro 2a gives high yield of the products compared to those of the other substituents such as 2b-3j.
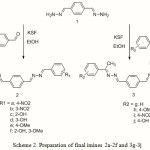 |
Scheme 2: Preparation of final imines 2a-2f and 3g-3j |
Synthesis of the final products 2a-2f and 3g-3j, in the absence of montmorillonite KSF either failed or completed in a long reaction time together with several byproducts. Performing a reaction in the presence of con. HCl as a catalyst due to conversion of some of the amine to its related hydrochloride salt gave only a tiny amount of mixture of salty products. While, we substitute montmorillonite KSF clay as a solid catalyst instead of the con. HCl surprisingly we covered a good yield of the final Schiff base products.
The UV-Vis spectra of compounds 2a-2f and 3g-3j compare to the UV-Vis spectra of the initial imine 1 have been recorded in the DMF solvent in the wavelength rang 800-250 nm (Figure 1 & 2-Table 1). Figure1 & 2 indicate that the products 2a-2f and 3g-3j compare to the initial imine 1 (λ max= 334.5) except to the 2d that shifted to the blue the other compounds all are shifted to the red field. This red shift are attributed to the long rang π→π* transitions of the respective compounds in the UV region. Absorption spectra of the prepared Schiff bases, with various electron donating and withdrawing abilities, in DMF, are shown in Figure1 It can be found that when introducing the nitro group in the aryl component of the Schiff base, 2a and 2c, the absorption spectra give a remarkable less red shift than others. Compound 2a which has nitro group in para position compared to meta 2b nitro group, the absorption spectra give a remarkable red shift.
Table 1: Results of spectroscopic studies in DMF as solvent.
| Product | 1 | 2a | 2b | 2c | 2d | 2e | 2f | 3g | 3h | 3i | 3j |
| λ max | 334.5 | 347.4 | 348.5 | 375.2 | 359, 323.5 | 369.2, 358.8 | 363.5 | 359 | 373.5 | 357.5 | 357.5 |
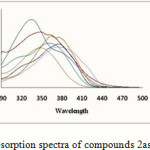 |
Figure 1: Absorption spectra of compounds 2as-2f in DMF Click here to View figure |
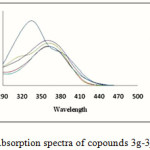 |
Figure 2: Absorption spectra of copounds 3g-3j in DMF. |
The UV-Vis spectra of compounds 2e, 3h in three different organic solvents with different polarities (Figure 3) reveal that the positions of the bands are influenced by the polarity of the solvents for 2e in DMF with highest polarity (λmax/ 369.2 nm), in a nonpolar solvent, CHCl3 (λmax/ 358 nm) and in EtOAc (λmax/ 354.8 nm) respectively were recorded. This solvochromic effect is attributed to the polarity of the solvents. In contrast to 2e, the 3h indicate a different absorption in CHCl3 (λmax/ 370.4 nm), in DMF (λmax/ 358nm) and EtOAc (λmax/ 359.2 nm) respectively was recorded.
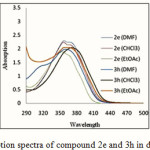 |
Figure 3: Absorption spectra of compound 2e and 3h in different solvents. |
The reaction of copper (II) acetate salt with the Schiff base 2c yields crystalline complex 4.
In the IR spectrum of compound 2c, a broad band observed in the region 3400-3100 cm-1 can be assigned to the ν (-OH) group. The absence of OH stretching band absorption in the IR spectra of complex 4 indicating that the oxygen atom is coordination to the copper (II) ion. In the other hand, the photometric study of the complex 4 indicate a ratio of ligand to copper ion as to be 1:1 this stoichiometric ratio allowed us to proposed the depicted structure (Scheme 3).
![Scheme 3: MM2 model of 4 shows [7,7] paracyclophane .](http://www.orientjchem.org/wp-content/uploads/2011/06/Vol27_Iss2_Nos_Syn_Sch3-150x150.jpg) |
Scheme 3: MM2 model of 4 shows [7,7] paracyclophane . |
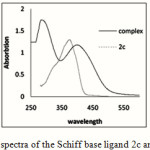 |
Figure 4: UV-Vis spectra of the Schiff base ligand 2c and its complex 4. |
The UV-Vis spectra of the complex 4 shows intense band in the range of 398.5, 310 and 330 nm relative to that of initial 2c ligand 375.2 nm . This different absorption behavior is assigned to the π→π* charge transfer band gap for complex 4 (Figure 4).
Acknowledgment
This study was supported in part bt Research Committee of the University of Guilan.
References
- Panneerselvam P., Nair R. R., Vijayalakshmi G., Subramania E. H., Sridhar S. K., Eur. J. Med. Chem., 40, 225(2005).
- Holla B. S., Veerandra B., Shivanada M. K., Poojary P., Eur. J. Med. Chem., 38, 759(2002).
- Pandeya S. N., Sriram D., Nath G., DeClercq E., Eur. J. Pharmacol. Sci., 9, 25(1999).
- Samadhiya S. and Halve A., Orient. J. Chem., 17, 119(2002).
- Cozzi P. G., Chem. Soc. Rev., 33, 410(2004).
- Li T. S. and Lin T. S., Chin. J. Org. Chem., 16, 385(1996).
- Zare L., Mahmoodi N. O., Yahyazadeh A., Mamaghani M., Tabatabaeian K., Chin. Chem. Lett., 21, 538(2010).
- Habibi D., Mahmoodi N., Marvi O., Can. J. Chem., 85, 81(2007).
- Habibi D., Mahmoodi N., Marvi O., Synth. Commun., 37, 3165(2007).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


