Ft-Ir and Computational Study of Sulphaguanidine
Asha Chandran1, Hema Tresavarghese2, C. Yohannan Panicker3, Rajendran4
1Department of Chemistry, TKM College of Arts and Science, Kollam, Kerala India. 2Department of Physics, Fatima Mata National College, Kollam, Kerala India. 3Department of Physics, TKM College of Arts and Science, Kollam, Kerala India. 4Department of Chemistry, University College, Trivandrum Kerala India.
The vibrational wavenumbers of sulphaguanidine were calculated using Gaussian03 software at different levels and compared with experimentally observed data. The predicted infrared intensities, Raman activities and first hyperpolarizability are reported. The calculated geometrical parameters (DFT) are in agreement with that of similar derivatives. The potential energy scan studies for different torsion angles are also reported. The splitting of NH stretching wavenumber in the infrared spectrum indicates the weakening of the NH bond.
KEYWORDS:FT-IR; Computation; Sulphaguanidine
Download this article as:| Copy the following to cite this article: Chandran A, Tresavarghese H, Panicker C. Y, Rajendran R. Ft-Ir and Computational Study of Sulphaguanidine. Orient J Chem 2011;27(2). |
| Copy the following to cite this URL: Chandran A, Tresavarghese H, Panicker C. Y, Rajendran R. Ft-Ir and Computational Study of Sulphaguanidine. Orient J Chem 2011;27(2). Available from: http://www.orientjchem.org/?p=25001 |
Introduction
Sulfonamides form a significant class of compounds in medicinal and pharmaceutical chemistry with several biological applications1-4. There has been growing interest in using organic materials for nonlinear optical (NLO) devices, functioning as second harmonic generators, frequency converters, electro-optical modulators, etc. because of the large second order electric susceptibilities of organic materials. The organic compound showing high hyperpolarizability are those containing an electron-donating group and an electron withdrawing group interacting through a system of conjugated double bonds. In the case of sulfonamides, the electron withdrawing group is the sulfonyl group5. To our knowledge, no theoretical HF or density functional theory (DFT) calculations, or detailed vibrational infrared analyses, have been performed on the title compound.
Experimental
All the chemicals were procured from Sigma-Aldrich, USA. The FT-IR spectrum was recorded using a Bruker IFS 28 spectrometer in KBr pellets, number of scans 16, resolution 2 cm-1.
Computational details
Calculations of the title compound were carried out with Gaussian03 software program6 using the HF/6-31G* and B3LYP/6-31G* basis sets to predict the molecular structure and vibrational wavenumbers. The DFT hybrid B3LYP functional tends also to overestimate the fundamental modes; therefore scaling factors have to be used for obtaining a considerably better agreement with experimental data. Therefore, a scaling factor of 0.9613 and 0.8929 were uniformly applied to the DFT and HF calculated wavenumbers7. Potential energy surface scan studies have been carried out to understand the stability of planar and non-planar structures of the molecule. The profiles of potential energy surface for torsion angles N20-C19-N17-S14, N22-C19-N17-S14 and C19-N17-S14-C3 are calculated. The energy is minimum for -143.5° (-1040.33670), 38.4° (-1040.33511) and -172.8° (-1040.33577 Hartree) for the above torsion angles. The assignment of the calculated wavenumbers is aided by the animation option of MOLEKEL program, which gives a visual presentation of the vibrational modes8,9.
Results and Discussion
The observed IR bands with their relative intensities and calculated (scaled) wavenumbers and assignments are given in Table 1. The anti-symmetric and symmetric stretching modes of SO2 group appear in the region 1360-1310 and 1165-1135 cm-1, respectively10. The observed bands at 1305, 1130 cm-1 in the IR spectrum were assigned to the SO2 stretching modes. The DFT calculations give the anti-symmetric and symmetric stretching modes at 1314, 1135 cm-1. Although the region of the SO2 scissors (560 ± 40 cm-1) and that of SO2 wagging vibration (500 ± 55 cm-1) partly overlap, the two vibrations appear separately10. The scissoring mode is observed at 555 cm-1 in the IR spectrum. The wagging mode is observed at 511 cm-1 in the IR spectrum. The DFT calculations give these modes at 538 and 508 cm-1. Hangen et al.11 reported the SO2 stretching vibrations at 1314, 1308, 1274, 1157, 1147, 1133 cm-1 and SN stretching modes at 917, 920, 932, 948 cm-1 for sulfonamide derivatives. Chohan et al.12 reported SO2 stretching modes at 1345, 1110 cm-1 and SN and CS stretching modes at 833 cm-1 for sulfonamide derivatives. The twisting SO2 mode is expected10 in the region 440 ± 50 cm-1 and the rocking mode at around 350 cm-1. For the title compound, the band observed at 412 cm-1 in the IR spectrum is assigned as the twisting mode of SO2 moiety. The DFT calculations give these modes at 416 and 399 cm-1. Rodriguez et al.13 reported the SO2 bands in the range 1242-1394, 590-632 and 460-470 cm-1. The SN stretching vibration10 is expected in the region 905 ± 30 cm-1. The band calculated at 830 cm-1 is assigned as SN stretching mode and the band at 849 cm-1 in the IR spectrum is assigned as this mode. The C-S stretching mode is observed at 658 cm-1 (DFT)10.
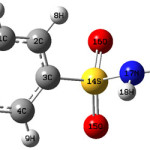 |
Scheme 1 Click here to View scheme |
The N-H stretching vibrations generally give rise to bands14,15 at 3500-3300 cm-1. The NH stretching bands are observed at 3388, 3346 cm-1 in the IR spectrum and at 33357, 3340 cm-1 (DFT) theoretically. In the present study, the NH stretching band has split into a doublet, 3346 and 3215 cm-1 in the IR spectrum owing to the Davydov coupling between neighboring units. A similar type of splitting observed in acetanilide16,17 and N-methylacetamide18 in the stretching band is attributed to the Davydov splitting. The splitting of about 131 cm-1 in the IR spectrum is due to strong intermolecular hydrogen bonding. Furthermore, the N-H stretching wavenumbers are red shifted by 125 cm-1 in the IR spectrum with a strong intensity from the computed wavenumbers, which indicates the weakening of the N-H bond19. In N-monosubstituted amides, the in-plane bending frequency and the resonance stiffened C-N band stretching frequency fall close together and therefore interact. The NH deformation band of guanidine structural motif is expected in the region10,20 1395 ± 25 cm-1. The DFT calculations give this modes at 1394 and 1348 cm-1. The out-of-plane NH deformation is expected in the region10 650 ± 50 cm-1 and bands at 634 and 617 cm-1 (DFT) are assigned as this mode. Wang and Ma21 reported NH stretching bands in the region, 3365-3374 cm-1 and guanidine C=N stretching bands in the region 1598-1638 cm-1. According to Henry et al.22 IR spectrum exhibits typical features for Schiff base possessing several bands of υNH vibration with maxima in the range 3380-3270 cm-1 and bands due to υC=N at 1635 cm-1. The C=N stretching skeletal bands23-25 were observed in the range 1627-1566 cm-1. DFT calculations give the υC=N mode at 1649 cm-1. The C-N stretching vibration10 is moderately to strongly active in the region 1275 ± 55 cm-1. Primary aromatic amines with nitrogen directly on the ring absorb at 1330-1200 cm-1 because of the stretching of the phenyl C-N bond20. For the title compound, the C6-N11 stretching mode is observed at 1236 cm-1 in the IR spectrum and at 1285 cm-1 theoretically. Panicker et al.26 reported CN stretching mode at 1219, 1237 (IR), 1222 (Raman) and at 1292, 1234, 1200 cm-1 theoretically. The CN stretching modes C19-N17 and C19-N22 are assigned at 1050, 1047 cm-1 theoretically, and at 1050 cm-1 in the IR spectrum, which is expected10 in the range 950-1115 cm-1. Fanchiang and Tseng27 reported CN stretching modes at 1037 and 1120 cm-1.
The NH2 stretching modes of guanidine10 are expected in the region 3260-3390 cm-1 and in the present case bands observed at 3496, 3438 cm-1 in the IR spectrum and bands at 3617, 3585, 3497, 3407 cm-1 (DFT) are assigned as NH2 stretching modes. Topacli and Topacli28 reported the calculated wavenumbers in the range 3670-3920 cm-1 for NH2 stretching modes. The bands corresponding to the δNH2 vibrations10 are expected in the region 1610 ± 30 cm-1. In the IR spectrum δNH2 is observed at 1620 cm-1. The calculated value are 1649, 1642 cm-1. The rocking/twisting mode of NH2 is expected in the region 1195 ± 90 cm-1 and the DFT calculations give this mode at 1187 and 1111 cm-1. Experimentally, bands are observed at 1176 and 1111 in the IR spectrum. The wagging mode of NH2 is expected10 in the range 840 ± 55 cm-1 and is observed at 747 cm-1 in the IR spectrum. The DFT calculations give these modes at 769 and 731 cm-1. The torsion NH2 mode is expected in the range 355 ± 65 cm-1 and the bands at 340, 307 cm-1 (DFT) are assigned as this mode. For sulfonamide derivatives, the NH2 modes are reported at29 3390, 3395, 3399 cm-1 and NH modes at 3253, 3230, 3255 cm-1.
The aromatic CH stretching vibrations10 absorb weakly to moderately between 3120-3000 cm-1. The B3LYP calculations give bands in the range 3081-3116 cm-1 as υCH stretching modes. Experimentally, no bands are observed. The benzene ring possesses six ring stretching vibrations of which the four with the highest wavenumbers occurring near 1600, 1580, 1490 and 1440 cm-1 are good group vibrations10. The fifth ring stretching vibration is active near 1315 ± 65 cm-1, a region that overlaps strongly with that of the CH in-plane deformation10. The sixth ring stretching vibration, the ring breathing mode, appears as a weak band near 1000 cm-1 in mono-, 1,3-di, and 1,3,5-trisubstituted benzenes. In the otherwise substituted benzenes, however, this vibration is substituent sensitive and difficult to distinguish from other modes. The ring breathing mode for the para-substituted benzenes with entirely different substituents30 has been reported to be strongly IR active with typical bands in the interval 780-840 cm-1. For the title compound, this is confirmed by the band in the IR spectrum at 823 cm-1, which finds support from the computational results (820 cm-1 by DFT). The ring breathing mode of para-substituted benzenes are reported at 804 and 792 cm-1 experimentally and at 782 and 795 cm-1 theoretically31,32. For the title compound, the phenyl ring stretching modes are observed at 1572, 1534, 1492, 1434 cm-1 in the IR spectrum and at 1593, 1568, 1496, 1431, 1321 cm-1 theoretically. For para-substituted benzenes, the δCH modes10 are seen in the range 995-1315 cm-1 and in the present case the bands observed at 1176, 1086 cm-1 in the IR spectrum and at 1321, 1187, 1111, 1027 cm-1 (B3LYP) are assigned as these modes. The CH out-of-plane deformations10 are observed between 1000 and 700 cm-1. Generally, the CH out-of-plane deformations with the highest wavenumbers are weaker than those absorbing at lower wavenumbers. These γCH modes are observed at 1010, 941 cm-1 in the IR spectrum and at 992, 946, 811, 807 cm-1 theoretically. The strong CH out-of-plane deformation band occurring at 840 ±50 cm-1 is typical for 1,4-di-substituted benzenes10. For the title compound, a band is observed at 810 cm-1 in the IR spectrum. Again according to literature10,20 a lower γCH absorbs in the neighborhood 820 ± 45 cm-1, but is much weaker or infrared inactive. The DFT calculations give a γCH at 807 cm-1 and no band is experimentally observed for this mode. The substituent sensitive modes of the phenyl ring are also identified and assigned.
The geometrical parameters given by the theoretical results obtained are almost comparable with the reported structural parameters of similar derivatives. For benzene sulfonamide derivatives, Loughrey et al.33 reported the bond lengths, S14-O15=1.4337, S14-O16=1.4256, S14-N17=1.6051, S14-C3=1.7737, C6-N11=1.4212Å, whereas the corresponding values in the present case are, 1.6376, 1.6482, 1.8364, 1.8463, 1.3747Å. For the title compound the DFT calculations give the bond angles C3-S14-O15=108.1, C3-S14-O16=111.6, S14-N17-H18=111.8, O15-S14-O16=118.3, O15-S14-N17=114.3, O16-S14-N17= 104.0, N17-S14-C3=98.9, C6-N11-H12,13=121.1, 121.2, S14-C3-C4=118.1, S14-C3-C2=119.4, C4-C3-C2=122.5, C3-C4-C5=118.7, C4-C5-C6=120.6, C5-C6-C1=119.0, N11-C6-C1=120.5, N11-C6-C5=120.6, C6-C1-C2=120.6°. The corresponding reported values are 106.5, 107.4, 110.0, 119.5, 106.1, 107.7, 109.3, 118.8, 120.5, 119.1, 120.4, 119.9, 120.3, 119.3, 122.4, 118.2, 120.4°33. Loughrey et al.33 reported the torsion angles O15-S14-C3-C4, O15-S14-C3-C2, O16-S14-C3-C4, O16-S14-C3-C2, N17-S14-C3-C4, N17-S14-C3-C2, S14-C3-C4-C5, C2-C3-C4-C5, S14-C3-C2-C1, C4-C5-C6-N11, N11-C6-C1-C2, as -142.0, 38.5, -12.8, 167.7, 103.8, -75.7, 143.9, -39.2, 177.6, -179.9, -0.4, 179.2, 178.1, -178.6, whereas the corresponding values for the title compound are, -0.0, -177.0, 131.7, -46.0, -119.3, 63.1, -178.8, -1.2, 178.8, -180.0, -180.0°. Petrov et al.34 reported the molecular structure and conformations of benzenesulfonamide by gas electron diffraction and quantum chemical calculations and according to their results, the bond lengths, CS, SN, SO vary in the range 1.7756-1.793, 1.663-1.6925, 1.4284-1.445Å, the bond angles, CSN, CSO, NSO, HNS, HNH, vary in the range, 103.9-107.1, 107.6-107.8, 105.5-107.7, 111-113.7, 112.6-113.6°. These values are in agreement with the corresponding values for the title compound. Labisbal et al.35 reported the bond lengths, SO=1.4269-1429, SN=1.6202, SC=1.7582, N22-C19=1.4103, N20-C19=1.2723, N17-C19=1.3483Å whereas the corresponding values for the title compound are 1.6376-1.6482, 1.8364, 1.8463, 1.3688, 1.2924, 1.4238Å. The values of bond angles O15-S14-O16=118.6, O16-S14-N17=108.9, O15-S14-N17=104.9, O15,16-S14-C3 =107.9-108.3, N17-S14-C3 =107.9, C19-N17-S14 =123.0° reported by Labisbal et al.35 are in agreement with the corresponding values of the title compound.
The calculated first hyperpolarizability of the title compound is 6.87 × 10-30 esu, which comparable with the reported values of similar derivatives, but experimental evaluation of this data is not readily available. Kucharski et al.36 reported the first hyperpolarizability of certain sulfonamide amphiphiles by calculation and hyper-Rayleigh scattering in the range 0.2156-0.189 × 10-30 esu. We conclude that the title compound is an attractive object for future studies of nonlinear optical properties.
Table 1: Calculated vibrational wavenumbers (scaled), measured infrared band positions and assignments
|
HF/6-31G* |
B3LYP/6-31G* | υ(IR) (cm-1) | Assignments | ||||
|
υ (cm-1) |
IR Intensity |
Raman Activity |
υ
(cm-1) |
IR Intensity | Raman Activity | ||
|
3568 |
44.72 | 60.76 | 3617 | 27.03 | 74.80 | υasNH2 | |
| 3543 | 99.63 | 58.03 | 3585 | 66.34 | 84.90 | υasNH2 | |
| 3450 | 104.88 | 184.30 | 3497 | 99.88 | 268.15 | 3496 s | υsNH2 |
| 3429 | 87.24 | 34.56 | 3407 | 44.79 | 52.82 | 3438 s | υsNH2 |
| 3381 | 118.79 | 99.42 | 3357 | 119.39 | 200.96 | 3388 s | υNH |
| 3364 | 16.32 | 243.51 | 3340 | 11.19 | 355.37 | 3346 s, 3215 m | υNH |
| 3044 | 7.14 | 78.40 | 3116 | 3.44 | 80.53 | υCH | |
| 3039 | 1.86 | 62.50 | 3113 | 7.18 | 55.44 | υCH | |
| 3015 | 11.45 | 84.85 | 3082 | 14.13 | 110.37 | υCH | |
| 3014 | 11.19 | 81.51 | 3081 | 13.38 | 96.72 | υCH | |
| 1681 | 563.53 | 23.40 | 1659 | 306.24 | 70.27 | υCN | |
| 1661 | 167.54 | 10.96 | 1649 | 146.95 | 18.39 | δNH2 | |
| 1655 | 239.81 | 20.12 | 1642 | 229.57 | 51.47 | 1620 s | δNH2 |
| 1599 | 251.27 | 112.49 | 1593 | 219.96 | 97.04 | 1572 m | υPh |
| 1579 | 22.87 | 1.90 | 1568 | 7.54 | 1.07 | 1534 s | υPh |
| 1505 | 139.84 | 1.52 | 1496 | 70.25 | 13.79 | 1492 m | υPh |
| 1437 | 10.11 | 0.58 | 1431 | 5.69 | 0.62 | 1434 w | υPh |
| 1411 | 275.69 | 2.19 | 1394 | 163.13 | 23.02 | δNH | |
| 1365 | 67.23 | 4.37 | 1348 | 1.18 | 0.57 | δNH | |
| 1333 | 9.29 | 0.70 | 1321 | 94.38 | 2.52 | υPh,
δCH |
|
| 1300 | 152.82 | 11.46 | 1314 | 20.27 | 1.21 | 1305 w | υasSO2 |
| 1253 | 2.68 | 0.29 | 1285 | 75.74 | 11.24 | 1236 s | υC6-N11 |
| 1188 | 69.54 | 4.45 | 1187 | 51.30 | 3.45 | 1176 s | ρNH2,
δCH |
| 1130 | 6.34 | 0.37 | 1135 | 9.48 | 1.14 | 1130 s | υsSO2 |
| 1124 | 17.94 | 5.68 | 1111 | 14.92 | 12.70 | 1086 s | ρNH2,
δCH |
| 1084 | 277.03 | 7.94 | 1050 | 39.32 | 17.81 | 1050 s | υC19-N17 |
| 1062 | 62.78 | 27.06 | 1047 | 265.07 | 37.39 | υC19-N22 | |
| 1047 | 3.62 | 1.40 | 1027 | 1.68 | 0.18 | δCH | |
| 1033 | 2.07 | 0.15 | 992 | 5.13 | 10.28 | 1010 w | γCH |
| 1024 | 5.44 | 0.52 | 973 | 0.07 | 0.20 | 964 w | δSNC |
| 998 | 3.48 | 3.78 | 946 | 1.27 | 0.17 | 941 w | γCH |
| 965 | 139.32 | 8.60 | 918 | 106.70 | 25.93 | γCH | |
| 873 | 136.53 | 1.05 | 900 | 63.60 | 5.55 | δPh(X) | |
| 851 | 3.69 | 3.39 | 830 | 41.17 | 1.68 | 849 w | υSN |
| 832 | 21.18 | 15.70 | 820 | 77.02 | 16.44 | 823 s | Ring breathing |
| 810 | 5.71 | 24.97 | 811 | 17.33 | 19.57 | 810 s | γCH |
| 802 | 73.79 | 7.32 | 807 | 20.43 | 4.28 | γCH | |
| 786 | 229.22 | 9.49 | 769 | 61.61 | 18.34 | ωNH2 | |
| 749 | 133.37 | 8.64 | 731 | 170.92 | 62.87 | 747 w | ωNH2 |
| 735 | 16.28 | 1.21 | 703 | 2.70 | 1.98 | δPh(X) | |
| 679 | 26.63 | 12.39 | 689 | 44.05 | 33.06 | 687 s | γPh |
| 653 | 173.70 | 1.73 | 658 | 183.66 | 12.91 | υCS | |
| 638 | 0.32 | 6.31 | 634 | 0.86 | 4.81 | γNH | |
| 616 | 27.10 | 7.54 | 617 | 28.48 | 7.05 | 604 s | γNH |
| 563 | 37.04 | 20.86 | 538 | 39.69 | 64.53 | 555 s | δSO2 |
| 540 | 39.39 | 0.79 | 524 | 83.61 | 0.99 | 542s | δPh |
| 526 | 159.86 | 1.81 | 508 | 79.71 | 2.09 | 511 w | ωSO2 |
| 486 | 407.94 | 6.72 | 464 | 282.26 | 26.97 | 463 w | γPh(X) |
| 468 | 207.88 | 1.77 | 442 | 230.46 | 4.70 | δPh(X) | |
| 443 | 29.27 | 4.17 | 426 | 70.48 | 9.52 | γPh | |
| 425 | 1.30 | 0.20 | 416 | 1.99 | 1.14 | 412 w | τSO2 |
| 410 | 2.20 | 2.13 | 407 | 11.70 | 3.41 | δCX(X) | |
| 385 | 12.53 | 6.62 | 401 | 0.48 | 2.04 | δCX(X) | |
| 377 | 0.26 | 1.86 | 399 | 9.02 | 8.38 | ρSO2 | |
| 351 | 9.52 | 4.76 | 340 | 10.41 | 10.82 | ρNH2 | |
| 332 | 4.92 | 2.57 | 307 | 5.10 | 30.45 | ρNH2 | |
| 288 | 22.40 | 6.41 | 285 | 9.22 | 11.57 | γCX(X) | |
| 262 | 3.50 | 2.22 | 246 | 5.01 | 9.35 | δCNS | |
| 241 | 46.89 | 6.69 | 238 | 30.01 | 7.56 | γCNS | |
| 222 | 2.79 | 6.90 | 215 | 1.43 | 13.32 | γCX(X) | |
| 149 | 1.84 | 0.57 | 144 | 3.71 | 0.59 | δNC=N | |
| 140 | 2.93 | 2.74 | 136 | 0.49 | 3.81 | tNH2 | |
| 91 | 1.28 | 2.60 | 92 | 0.43 | 3.12 | tNH2 | |
| 60 | 3.17 | 4.31 | 58 | 3.23 | 2.77 | tNCNH2 | |
| 51 | 2.16 | 5.21 | 51 | 2.00 | 9.48 | tPh | |
| 30 | 0.52 | 9.66 | 28 | 0.35 | 11.60 |
tSO2 |
|
υ-stretching, δ-in-plane bending, γ-out-of-plane bending, τ-twist, t- torsion; Ph-phenyl ring; X-substituent sensitive; subscripts; as, asymmetric; s, symmetric.
References
- Tilles, S.A., Southern Med. J. 94: 817 (2001).
- Slatore, C.G., and Tilles, S., Immun. Allergy. Clinics North Am. 24: 477 (2004).
- Brackett, C.C., Singh, H.,and Block, J.H., Pharmacotherapy 24: 856 (2004).
- Eroglu, E., Int. J. Mol. Sci. 9: 181 (2008).
- Liu, X., Liu, L., Lu, X., Zheng, J., Wang, W., and Fang, Y., Thin Solid Films 217: 174 (1992).
- Frisch, M.J., et al. Gaussian 03, Revision C.02 Gaussian, Inc., Wallingford CT(2004).
- Foresman, J.B., in: Frisch, E., (Ed.), Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian”, Pittsburg, PA, (1996).
- Flukiger, P., Luthi, H.P., Portmann, S., and Weber, J., MOLEKEL 4.3, Swiss Centre for Scientific Computing, Manno, Switzerland (2000-2002).
- Portmann, S., and Luthi, H.P., Chimia 54: 766 (2000).
- Roeges, N.G.P., A Guide to the Complete Interpretation of the Infrared spectra of organic structures, Wiley, NewYork (1994).
- Hangen, A., Bodoki, A., Opren, L., Alznet, G., Liu-Gonzalez, M., and Borras, J., Polyhedron 29: 1305 (2010).
- Chohan, Z.H., Youssoufi, M.H., Jarrahpour, A., and Hadda, T.B., Eur. J. Med. Chem. 45: 1189 (2010).
- Rodriguez, A., Sanchez-Vergara, M.E., Garcia-Montalvo, V., Ortiz-Rebollo, A., Alvarez-Bada, J.R., and Alvarez-Toledano, C., Spectrochim. Acta 75: 479 (2010).
- Bellamy, L.J., The IR spectra of Complex Molecules, John Wiley and Sons, New York (1975).
- Spire, A., Barthes, M., Kallouai, H., and De Nunzio, G., Physics D 137: 392 (2000).
- Edler, J., Pfister, R., Pouthier, V., Falvo, C., and Hamm, P., Phys. Rev. Lett. 93: 106405 (2004).
- Edler, J., Hamm, P., and Scott, A.C., Phys. Rev. Lett. 88: 067403 (2002).
- Edler, J., and Hamm, P., Phys. Rev. B69: 214301 (2004).
- Barthes, M., De Nunzio, G., and Ribet, G., Synth. Met. 76: 337 (1996).
- Colthup, N.B., Daly, L.H., and Wiberly, S.E., Introduction to Infrared and Raman Spectroscopy, Academic Press, New York (1975).
- Wang, B., and Ma, H.Z., Inorg. Chem. Commun. 4: 248 (2001).
- Henry, N., Lagrenee, M., and Abraham, F., Inorg. Chem. Commun. 11: 1071 (2008).
- Yalcin, I., Sener, E., Ozden, O., and Akin, A., Eur. J. Med. Chem. 25: 705 (1990).
- Saxena, R., Kaudpal, L.D., and Mathur, G.N., J. Polym. Sci. Part A: Polym. Chem. 40: 3959 (2002).
- Silverstein, R.M., and Webster, F.X., Spectrometric Identification of Organic Compounds, ed. 6, Wiley, Singapore (2003).
- Panicker, C.Y., Varghese, H.T., Mariamma, K.C., John, K., Mathew, S., Vinsova, J., Van Alsenoy, C., and Mary, Y.S., J. Raman Spectrosc. 41: 707 (2010).
- Fanchiang, J.M., and Tseng, D.H., Chemosphere, 77: 214 (2009).
- Topacli, C., Topacli, A., J. Mol. Struct. 644: 145 (2003).
- Chohan, Z.H., Youssoufi, M.H., Jarrahpour, A., and Hadda, T.B., Eur. J. Med. Chem. 45: 1189 (2010).
- Varsanyi, G., Assignments of Vibrational Spectra of Seven Hundred Benzene Derivatives, Wiley, New York 1974.
- Mary, Y.S., Varghese, H.T., Panicker, C.Y., Ertan, T., Yildiz, I., and Temiz-Arpaci, O., Spectrochim. Acta 71: 566 (2008).
- Ambujakshan, A.R., Madhavan, V.S., Varghese, H.T., Panicker, C.Y., Temiz-Arpaci, O., Tekiner-Gulbas, B., and Yildiz, I., Spectrochim. Acta 69: 782 (2007).
- Loughrey, B.T., Williams, M.L., and Healy, P.C., Acta Cryst. E65: o2087 (2009).
- Petrov, V., Petrova, V., Girichev, G.V., Oberhammer, H., Giricheva, N.I., and Ivanor, S., J. Org. Chem. 71: 2952 (2006).
- Labisbal, E., Rodriguez, L., Sousa-Pedrares, A., Alonso, M., Vizoso, A., Romero, J., Garcia-Vazquez, J.A., and Sousa, A., J. Organomet. Chem. 691: 1321 (2006).
- Kucharski, S., Janik, R., and Katz, P., J. Mater. Chem. 9: 395 (1999).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


