Synthesis of 1-azabenzo[a]phenoxazin-5-one and 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one and their functionalized aryl derivatives via Mizoroki-Heck arylation methodology
Oni Timothy Toba, Okoro Uchechukwu Chris and Ugwu David Izuchukwu*
Department of Pure and Industrial Chemistry, University of Nigeria
DOI : http://dx.doi.org/10.13005/ojc/310144
Article Received on :
Article Accepted on :
Article Published : 21 Feb 2015
The synthesis of angular 1-azabenzo[a]phenoxazin-5-one and 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one and their functionalized aryl derivatives via Mizoroki-Heck arylation methodology is reported. 11-Amino-1,8,10-triazabenzo[a]phenoxazin-5-one (16) and 1-azabenzo[a]phenoxazin-5-one (18) were synthesized by the reaction of 7-chloro-5,8-quinolinequinone (obtained by a multistage conversion of 8-hydroxyquinoline) with 4,5-diamino-6-hydroxypyrimidine and 2-aminophenol respectively in the presence of anhydrous sodium acetate. 11-Amino-1,8,10-triazabenzo[a]phenoxazin-5-one and 1-azabenzo[a]phenoxazin-5-one were subjected to Mizoroki-Heck coupling reaction by refluxing with iodobenzene derivatives, using 1,4-bis(diphenylphosphino)butane-palladium(11) chloride as catalyst, 1,4-bis-(2-hydroxy-3,5-ditert-butylbenzyl)piperazine as ligand and methanol as solvent at 60-650C for 4 h to afford in excellent yield aryl derivatives of angular 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one (19a-c) and 1-azabenzo[a]phenoxazin-5-one (20a-c) respectively. The structures were established by UV/visible, FTIR, proton-NMR and carbo-13 NMR spectral and elemental analysis.
KEYWORDS:1-azaphenoxazinone; 1;8;10-triazaphenoxazinobe; Mizoroki-Heck; arylation; synthesis
Download this article as:| Copy the following to cite this article: Toba O. T, Chris O. U, lzuchukwu u. d. Synthesis of 1-azabenzo[a]phenoxazin-5-one and 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one and their functionalized aryl derivatives via Mizoroki-Heck arylation methodology. Orient J Chem 2015;31(1). |
| Copy the following to cite this URL: Toba O. T, Chris O. U, lzuchukwu u. d. Synthesis of 1-azabenzo[a]phenoxazin-5-one and 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one and their functionalized aryl derivatives via Mizoroki-Heck arylation methodology. Orient J Chem 2015;31(1). Available from: http://www.orientjchem.org/?p=7333 |
Introduction
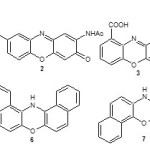
Phenoxazine derivatives are compounds of great interest because of their several successful applications as dyes and drugs. They exhibit strong biological activities ranging from antidepressant1, antitumour2, anticancer3, antibacterial4, antituberculosis5 and schizophrenia agents6. Among the several industrial applications of phenoxazine derivatives are their use as acid-base indicator7, biological stains8, laser dyes9 and chromophoric compounds10 in host guest artificial protonic antenna systems. Ruan et al11 has reported phenoxazinone derivatives of antitumour potential in wild-type and drug resistant tumor cells. Zaytsey et al12 reported the synthesis of chromogenic phenoxazinone substrate for β-alanyl amino peptidase. Abe et al13 has reported the synthesis of 2-amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazin-3-one that could prevent growth of human lung carcinoma cells and induce apoptosis. Gerasimova et al14 reported the synthesis of fluorinated benzophenoxazines via sequential SNAr substitution reactions of fluorinated aromatics. Jose and Burgess15 has also reported the synthesis of benzophenoxazine based fluorescent dyes used for labeling biomolecules. Carr et al16 reported plectosphaeroic acid, a marine fungal natural product containing phenoxazine ring as inhibitor of indoleamine 2,3-dioxygenase. Amino phenoxazinone core has also been reported in numbers of bioactive substances such as exfoliazone (1), the venezuelines (2), cinnabarinic acid (3), the chandrananimycin A or B (4) to mention but a few. Jeromin17 reported phenoxazine system derivatives which have been successfully applied for phenoxyl radical stabilization. Thimmaiah et al18 reported the synthesis of series of N10-substituted phenoxazines with Akt inhibitory potential. The structural modifications of parent phenoxazine ring were borne out of the need to minimize their undesirable effect and at the same time enhance the biological activities. Angular phenoxazines are phenoxazine derivatives that have non-linear arrangement of the ring system. Few examples of angular phenoxazine derivatives includes dibenzo[a,i]phenoxazine (5), dibenzo[a,j]phenoxazine (6), dibenzo[a,h]phenoxazine (7) and benzopyranol[3,4-b]benzoxazine (8) in which one of the ring carbon atom was replaced with oxygen.
 |
Scheme 1 Click here to View scheme |
Experimental
Reactions were carried out under nitrogen atmosphere where necessary. Melting points were determined with a Fischer Johns melting point apparatus and are uncorrected. UV/ visible spectra were recorded in acetone on a Unicon UV- 2500PC spectrophotometer using matched 1cm quartz cells, absorptions were measured in nanometer (nm). IR spectra were recorded on 8400s Fourier Transform Infrared (FTIR) spectrophotometer and are reported in wave numbers (cm-1). UV/visible and IR spectral analysis were done at the National Research Institute for Chemical Technology (NARICT), Zaria, Kaduna State, Nigeria. Nuclear magnetic resonance (1H NMR and 13C NMR) spectra were obtained using a Jeol 400 MHz spectrometer at Strathclyde University, Scotland. Chemical shifts are reported in (δ) scale. The elemental analysis was done on a Heraeus CHN-O rapid analyzer. All reagents used were of technical grade were purchased from Aldrich in sure-seal bottles and were used without further purifications.
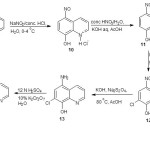
Synthesis of 8-hydroxy-5-nitrosoquinoline hydrochloride (10)
The method of Pratt and Drake19 was used to synthesize this compound. Into a liter beaker placed in an ice bath, a solution of 8-hydroxyquinoline (9) (58 g, 0.4 mol) and water (200 mL) was poured in, concentrated hydrochloric acid (75 mL) and ice (200 g) was added. Aqueous solution of sodium nitrite (NaNO2) (30 g) in water (100 mL) was added in portions to the mixture with vigorous stirring for over 1 h at 0-4 ºC. The mixture was allowed to stand overnight at 0ºC and the desired product filtered and washed with cold water. The product was air dried to afford a bright yellow solid. Yield 8.2 g, (92%), mp 181 ºC.
Synthesis of 8-hydroxy-5-nitroquinoline (11)
The oxidation of 8-hydroxy-5-nitrosoquinoline (10) to 8-hydroxy-5-nitroquinoline (11) was carried out according to the procedure of Petrow and Sturgeon20. Finely grounded 8-hydroxy-5-nitrosoquinoline hydrochloride (10) (15.0 g, 0.7 mmol) was added into a 500 mL beaker containing concentrated nitric acid (45 mL) and water (30 mL) in an ice bath. The mixture was stirred for 85 min at 17 °C. Equal volume of cold water was added and the mixture cooled to 0 °C and alkaline with cold concentrated potassium hydroxide solution pH 13.0. The red potassium salt was decomposed on neutralization with acetic acid, filtered by suction and washed with water. The residue was recrystallized from ethanol to afford a bright yellow crystal of 8-hydroxy-5-nitroquinoline. Yield 14.10 g (90.1%), mp 179 °C (lit 179.5-181.5 °C)20.
Synthesis of 7-chloro-8-hydroxy-5-nitroquinoline (12)
The chlorination of 8-hydroxy-5-nitroquinoline (11) was achieved using the literature20. 8-Hydroxy-5-nitroquinoline (11) (10 g) was suspended in water (1 L) in a two liter beaker and potassium hydroxide (1 M, 45 mL) was added. The mixture was stirred vigorously as sodium hypochlorite (72 mL) was added in portion for 90 min. After the addition of the hypochlorite, the mixture was stirred for further 2 h, neutralized with acetic acid and stirred to ensure complete conversion of the precipitate to free quinolinol. The mixture was then filtered and washed with water and the residue recrystallized from aqueous ethyl acetate to afford 7-chloro-8-hydroxy-5-nitroquinoline as a bright orange solid. Yield 8.50 g, (88%), mp 233-234 °C (lit 235 °C)20. The presence of the chloro group was confirmed using sodium fusion21.
Synthesis of 5-amino-7-chloro-8-hydroxy-5-nitroquinoline (13)
The reduction of 7-chloro-8-hydroxy-5-nitroquinoline (12) to 5-amino-7-chloro-8-hydroxy-5-nitroquinoline (13) was achieved using the literature20. 7-Chloro-8-hydroxy-5-nitroquinoline (12) (22.4 g, 0.1 mmol) was ground in a mortar with potassium hydroxide (1 M, 110 mL) to ensure complete reduction of the insoluble potassium salt. The suspension was transferred with the aid of water (280 mL) into a litre three necked round bottom flask equipped with a long magnetic stirring bar. The mixture was heated in a water bath at 50 ºC with vigorous stirring and potassium hydroxide (8 M, 70 mL) was added while heating continued. The mixture was treated with sodium dithionate (70 g). The mixture was reheated; maintained at 80 ºC for 10 min while a rapid stream of nitrogen gas was passed into the flask. After 10 min, more sodium dithionate (10 g) was added while the passage of nitrogen gas continued for another 10 min. The resulting suspension was cooled in ice under nitrogen gas and the precipitate filtered off by suction, washed with cold water containing trace of dithionate and dried rapidly in an oven to give 5-amino-7-chloro-8-hydroxyquinoline as a golden yellow solid. Yield 22.0 g (98 %), mp 171-172 °C (lit 173-174 °C)20.
7-Chloro-5,8-quinolinequinone (14)
7-Chloro-5,8-quinolinequinone (14) was synthesized by suspending 5-amino-7-chloro-8-hydroxyquinoline (13) (22.4 g, 0.1 mol) in water (600 mL) in a litre beaker equipped with a long magnetic stirring bar in an ice-salt bath. Sulphuric acid (6 M, 18 mL) was added to dissolve the amine, while vigorous stirring continued, the solution was cooled to 2 ºC and the salt precipitated out in a finely divided form. An ice-cold solution made of potassium dichromate (10%, 103 mL) and sulphuric acid (6 M, 71 mL) was then added. The mixture was stirred and cooled in the ice-salt bath for 15 min. The precipitated salt was filtered in cold buckner funnel containing trace of ice, washed with cold water and air dried to afford a light tan residue which was recrystallized from aqueous dimethyl formamide (DMF) to obtain 7-Chloro-5,8-quinolinequinone as a bright yellow solid. Yield 22.2 g (99%), mp 172-173 °C (lit 173.5-174.5 °C)20.
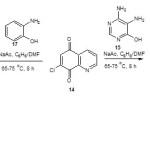
Synthesis of 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one (16): 4,5-Diamino-6-hydroxypyrimidine (15) (0.6 g, 0.005 mol), sodium acetate (1 g, 0.01 mol) and benzene (40 mL) mixed with DMF (5 mL) were charged into a 100 mL three necked round bottom flask fitted with short magnetic stirring bar and a reflux condenser. The mixture was stirred while heating on a water bath at 65-75 °C for 45 min. 7-Chloro-5,8-quinolinequinone (14) (0.9 g, 0.005 mol) was added and the stirring continued with heating at 70-75 °C for 8 h. The colour of the reaction mixture changed from light brown to greenish yellow, to red and intense red as the reaction progressed. After 8 h, the reaction mixture was filtered and the solvent allowed to evaporate to afford 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one (16) as intense reddish crystals. Yield 1.36 g, (90%), mp 287 °C. UV/vis (acetonelmax): 321.6 (log e 1.8135), 433.6 (log e 2.4450). FTIR (KBr, v, cm-1): 3420, 3289 (NH), 3090 (C-H aromatic), 1701 (C=O), 1630, 1603, 1570 (C=N), 1478 (C=C), 1245 (C-O), 1091 (C-N). 1H NMR (DMSO-d6, 400MHz) d: 7.2 (2H, d, J=8.90 Hz, Ar-H), 7.0 (1H, t, J= 7.09 Hz, Ar-H), 6.4 (1H, s, Ar-H), 6.3 (1H, s, 6-H), 5.4 (1H, s, NH2). 13C NMR (DMSO-d6, 400MHz) d: 165.40, 164.93, 162.34, 162.09, 159.90, 132.97, 132.08, 129.84, 127.29, 127.21, 122.04, 118.56, 110.67. C13H7N5O2 Calculated: C, 58.87, H, 2.66, N, 26.41, O, 12.06, found: C, 58.80, H, 2.70, N, 26.50, O, 12.02.
Synthesis of 1-azabenzo[a]phenoxazin-5-one (18)
2-Aminophenol (17) (0.5 g, 0.005 mmol), sodium acetate (1 g, 0.01 mol) and benzene (40 mL) mixed with dimethyl formamide (5 mL) were charged into a 100 mL three necked round bottom flask fitted with a magnetic stirring bar and a reflux condenser. The mixture was stirred while heating on a water bath at 70-75 ºC for 45 min. 7-Chloro-5,8-quinolinequinone (14) (0.9 g, 0.005 mol) was added and the stirring continued with heating at 70-75 °C for 8 h. The colour of the reaction mixture changed from yellowish ash to brown, to reddish brown and intense pinkish brown as the reaction progressed. After 8 h, the reaction mixture was filtered and the solvent evaporated from the filtrate to give 1-azabenzo[a]phenoxazin-5-one as intense brown solid. Yield 1.16 g, (87%), mp 272 °C. UV/vis (acetone)lmax: 321.4 (log e 1.8124), 436.8 (log e 2.4632). FTIR (KBr, v, cm-1): 2932 (C-H aromatic), 1586, 1583 (2C=N), 1488 (C=C), 1387, 1301 (2C-O), 1163 (C-N), 815, 752 (substitution in benzene). 1H NMR (DMSO-d6, 400MHz) d: 7.6 (m, 4H, Ar-H), 7.2 (m, 3H, Ar-H), 6.3 (1H, s, Ar-H). 13C NMR (DMSO-d6, 400MHz) d: 167.87, 159.98, 158.75, 145.65, 140.44, 129.43, 128.90, 128.32, 125.76, 122.32, 121.98, 121.09, 120.08, 120.01, 117.68. C15H8N2O2 calculated: C, 72.58, H, 3.25, N, 11.28, O, 12.89, found: C, 72.60, H, 3.20, N, 11.30, O, 12.90. The iodobenzene derivatives were prepared according to the method of Williamson22.
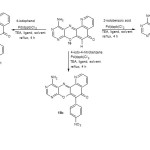
Synthesis of 11-Amino-1,8,10-Triazabenzo[A]Phenoxazin-5-One Angular Derivatives (19a-C)
In a two necked round bottom flask 0.5 g of 1,4-bis(2-hydroxy-3,5-di-tert-butylphenyl)piperazine ligand, 0.5 g of 1,4-bis(diphenylphosphino)butane palladium(II) chloride catalyst and 5 mL of methanol were placed with a magnetic stirring bar. After stirring for 5 min, 11-Amino-1,8,10-triazabenzo[a]phenoxazin-5-one (16) (1.3 g, 0.005 mol), iodobenzene derivatives (0.005 mol) and potassium carbonate (0.8 g) were added to the reaction flask. Stirring continued while heating on a water bath at 60-65 °C for 4 h. The colour of the reaction mixture changes from pinkish red to yellowish red and intense yellow as the reaction progressed. After 4 h, the reaction mixture was filtered and the filtrate allowed to evaporate to obtain the 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one angular derivatives in good to excellent yield.
6-(2-Carboxyphenyl)-11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one angular derivative (19a)
The compound was intense yellow, yield 0.7 g (56%), mp 295 °C. UV/vis (acetone) lmax: 322 (log e 1.8158), 436 (loge 2.4587), 496.2 (loge 2.7981), 546.4 (loge 3.0812). FTIR (KBr, v, cm-1): 3467 (NH), 1628 (C=O), 1281 (C-O). 1H NMR (DMSO-d6, 400MHz) d: 7.8 (4H, m, Ar-H), 6.9 (1H, s, Ar-H), 6.1 (3H, m, Ar-H), 5.8 (2H, s, NH2). 13C NMR (DMSO-d6, 400MHz) d: 171.09, 167.05, 162.89, 162.01, 160.32, 159.80, 142.01, 140.90, 140.34, 132.21, 132.10, 130.98, 130.05, 130.01, 128.92, 128.41, 125.55, 124.59, 124.07, 121.95. C20H11N5O4 calculated: C, 62.34, H, 2.88, N, 18.17, O, 16.61, found: C, 62.30, H, 2.90, N, 18.16, O, 16.61.
6-(4-nitrophenyl)-11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one angular derivative (19b)
The compound was brownish in colour, yield 1.0 g (87 %), mp 301 °C. UV/vis (acetone) lmax: 326.6 (loge 1.8417), 360 (loge 2.0301), 397.4 (loge 2.2410), 497 (loge 2.8027), 654.6 (loge 3.6914). FTIR (KBr, v, cm-1): 3366, 3258 (2NH), 3092 (C-H aromatic), 1710 (C=O), 1589 (C=N), 1494 (C=C), 1291 (C-O), 1169, 1094 (C-N), 841, 750 (substitution in benzene). 1H NMR (DMSO-d6, 400MHz) d: 7.2 (2H, d, J= 8.23 Hz, Ar-H), 7.0 (2H, d, J= 7.89 Hz, Ar-H), 6.7 (3H, m, Ar-H), 6.2 (1H, s, Ar-H). 13C NMR (DMSO-d6, 400MHz) d: 165.98, 162.09, 161.78, 160.60, 159.89, 159.09, 150.76, 150.53, 138.85, 135.32, 133.22, 133.01, 131.33, 130.92, 128.57, 127.06, 123.09, 120.70, 118.02. C19H10N6O4 calculated: C, 59.07, H, 2.61, N, 21.75, O, 16.57, found: C, 59.00, H, 2.60, N, 21.80, O, 16.60.
6-(4-nitrophenyl)-11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one angular derivative (19b)
The compound was brownish in colour, yield 1.0 g (87 %), mp 301 °C. UV/vis (acetone) lmax: 326.6 (loge 1.8417), 360 (loge 2.0301), 397.4 (loge 2.2410), 497 (loge 2.8027), 654.6 (loge 3.6914). FTIR (KBr, v, cm-1): 3366, 3258 (2NH), 3092 (C-H aromatic), 1710 (C=O), 1589 (C=N), 1494 (C=C), 1291 (C-O), 1169, 1094 (C-N), 841, 750 (substitution in benzene). 1H NMR (DMSO-d6, 400MHz) d: 7.2 (2H, d, J= 8.23 Hz, Ar-H), 7.0 (2H, d, J= 7.89 Hz, Ar-H), 6.7 (3H, m, Ar-H), 6.2 (1H, s, Ar-H). 13C NMR (DMSO-d6, 400MHz) d: 165.98, 162.09, 161.78, 160.60, 159.89, 159.09, 150.76, 150.53, 138.85, 135.32, 133.22, 133.01, 131.33, 130.92, 128.57, 127.06, 123.09, 120.70, 118.02. C19H10N6O4 calculated: C, 59.07, H, 2.61, N, 21.75, O, 16.57, found: C, 59.00, H, 2.60, N, 21.80, O, 16.60.
Synthesis of 1-azabenzo[a]phenoxazin-5-one angular derivatives (20a-c)
Into a two necked round bottom flask equipped with magnetic stirring bar, the mixture of 1,4-bis(2-hydroxy-3,5-di-tert-butylbenzyl)piperazine ligand (0.5 g), 1,4-bis(diphenylphosphino)butane palladium(II) chloride catalyst (0.5 g) and methanol (5 mL) were placed. After stirring for 5 min, 1-azabenzo[a]phenoxazin-5-one (18) (1.2 g, 0.005 mol), iodobenzene derivatives (0.005 mol) and potassium carbonate (0.8 g) were added to the reaction mixture, stirring continued while heating on a water bath at 60-65 ºC for 4 h, the colour of the reaction mixture changes from pinkish red to yellowish red and intense yellow as the reaction progressed. After 4 h, the reaction mixture was filtered and the filtrate was allowed to evaporate to dryness before extracting the product with acetone. The acetone extract was allowed to evaporate to obtain 1-azabenzo[a]phenoxazin-5-one angular derivatives in excellent yield.
6-(2-carboxyphenyl)-1-azabenzo[a]phenoxazin-5-one angular derivative (20a)
The compound was yellow in colour, yield 0.9 g (87%), mp 307 °C. UV/vis (acetone) lmax: 321.2 (loge 1.8113), 360 (loge 2.4722), 438.4 (loge 2.8162), 544.2 (loge 3.0688), 656 (loge 3.7004). FTIR (KBr, v, cm-1): 3473 (broad, OH), 2980 (C-H aromatic), 1576 (C=N), 1480 (C=C), 1288 (C-O), 1127 (C-N), 746 (substitution in benzene). 1H NMR (DMSO-d6, 400MHz) d: 7.3 (m, 4H, Ar-H), 6.8 (3H, m, Ar-H), 6.1 (4H, m, Ar-H). 13C NMR (DMSO-d6, 400 MHz) d: 171.00, 162.89, 155.80, 150.85, 145.01, 142.00, 140.84, 130.20, 128.41, 128.01, 126.67, 125.85, 125.59, 124.70, 123.95, 123.56, 123.05, 120.89, 120.45, 119.03, 117.65, 115.54. C22H12N2O4 calculated: C, 71.74, H, 3.28, N, 7.61, O, 17.37, found: C, 71.80, H, 3.30, N, 7.60, O, 17.30.
6-(4-nitrophenyl)-1-azabenzo[a]phenoxazin-5-one angular derivative (20b)
The compound was intense yellowish in colour, yield 1.1 g, (98%), mp 300 °C. UV/vis (acetone) lmax: 321.6 (loge 1.8135), 360 (loge 2.2151), 392.8 (loge 2.8117), 498.6 (loge 3.0812), 655.4 (loge 3.6959). FTIR (KBr, v, cm-1): 3100 (C-H aromatic), 1627 (C=O), 1282 (C-O). 1H NMR (DMSO-d6, 400MHz) d: 7.9 (2H, d, J= 9.04 Hz, Ar-H), 7.0 (2H, d, J= 6.89 Hz, Ar-H), 6.5 (3H, m, Ar-H), 6.1 (4H, m, Ar-H). 13C NMR (DMSO-d6, 400MHz) d: 169.11, 167.25, 160.22, 159.88, 132.23, 132.01, 130.88, 130.15, 130.06, 128.99, 128.11, 125.45, 124.99, 124.17, 121.55, 120.89, 120.04, 118.98, 116.27, 115.70, 110.01. C21H11N3O4 calculated: C, 68.29, H, 3.00, N, 11.38, O, 17.33, found: C, 68.30, H, 3.00, N, 11.36, O, 17.32.
6-(4-hydroxyphenyl)-1-azabenzo[a]phenoxazin-5-one angular derivative (20c)
The compound was ash in colour, yield 0.8 g (87%),mp 330 °C. UV/vis (acetone) λmax: 321.8 (loge 1.8147), 437.6 (loge 2.4677), 498.4 (loge 2.8105). FTIR (KBr, v, cm-1): 3436 (broad, OH), 2935 (C-H aromatic), 1581 (C=N), 1486 (C=C), 1288 (C-O), 1164 (C-N), 838, 758 (substitution in benzene). 1H NMR (DMSO-d6, 400MHz) d: 6.9 (2H, d, J= 8.74 Hz, Ar-H), 6.6 (2H, d, J= 7.88 Hz, Ar-H), 6.5 (2H, d, J= 8.93 Hz, Ar-H), 6.3 (1H, t, J= 8.04 Hz, Ar-H), 6.1 (4H, m, Ar-H). 13C NMR (DMSO-d6, 400MHz) d: 165.55, 159.22, 158.28, 132.32, 132.11, 130.88, 130.15, 128.90, 128.14, 125.44, 124.49, 124.18, 121.65, 120.88, 120.14, 118.78, 116.36, 115.08, 112.98, 112.09, 110.02. C21H12N2O3 calculated: C, 74.11, H, 3.55, N, 8.23, O, 14.10, found: C, 74.10, H, 3.52, N, 8.20, O, 14.15.
Result and Discussion
 |
Scheme 1 A Click here to View scheme |
 |
Scheme 2 Click here to View scheme |
 |
Scheme 3 Click here to View scheme |
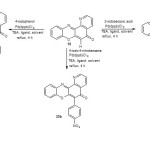 |
Scheme 4 Click here to View scheme |
The synthesis of 1-azabenzo[a]phenoxazin-5-one (18) was achieved by the reaction of 2-aminophenol (17) and 7-chloro-5,8-quinolinequinone (14) in the presence of sodium acetate at 70-75 °C for 8 h (scheme 2). The reaction of 1-azabenzo[a]phenoxazin-5-one (18) with iodobenzene derivatives in the presence of 1,4-bis(2-hydroxy-3,5-di-tert-butylbenzyl)piperazine as ligand, 1,4-bis(diphenylphosphino)butane palladium(II) chloride as catalyst, methanol as solvent and triethylamine as base at 60-65 °C under reflux for 4 h gave the aryl derivatives of angular 1-azabenzo[a]phenoxazin-5-one (20a-c) in excellent yield (scheme 4). 11-Amino-1,8,10-triazabenzo[a]phenoxazin-5-one (16) was synthesized by the reaction of 4,5-diamino-6-hydroxypyrimidine (15) and 7-chloro-5,8-quinolinequinone (14) in the presence of sodium acetate at 70-75 °C for 8 h using benzene/dimethyl formamide as solvent (scheme 2). The reaction of 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one (16) with iodobenzene derivatives in the presence of 1,4-bis(2-hydroxy-3,5-di-tert-butylbenzyl)piperazine as ligand, 1,4-bis(diphenylphosphino)butane palladium(II) chloride as catalyst, methanol as solvent and triethylamine as base at 60-65 °C under reflux for 4 h gave the aryl derivatives of angular 11-amino-1,8,10-triazabenzo[a]phenoxazin-5-one (19a-c) in excellent yield (scheme 3). The iodobenzene derivatives and 7-chloro-5,8-quinolinequinone (scheme 1) were prepared according to the literature and their purity ascertained using melting point. The synthesized monoaza and triazabenzo[a]phenoxazin-5-ones and their functionalized derivatives were characterized using UV/visible, FTIR, 1H NMR and 13C NMR spectroscopies and elemental analysis. The proposed structures of the novel compounds were in agreement with spectral and elemental analysis.
References
- Samet, A. V.; Kislyi, K. A.; Marshalkin, V. N.; Semenov, V. V. Izv. Ross. Akad. Nauk, Ser. Khim. 2006, 529
- Shimamoto, T.; Tomoda, A.; Ishida, R.; Ohyashiki, K. Am. Assoc. Can. Res. 2001, 7, 704-708.
- Horton, J. K.; Thimmaiah, K. N.; Harwood, F. C.; Kuttesch, J. F.; Houghton, P. J. Mol. Pharmacol. Abstract, 1993, 44, 552-559.
- Chu-Daniel, T. U. S. Chem. Abstr. 1986, 104, 109663K.
- Boothroyd, B.; Clark, E. R. J. Chem. Soc. 1952, 1499.
- Kapur, S.; McClelland, R. US patent 6890919, 2005.
- Mass, H.; Khatyr, A.; Calzaferri, G. Microporous and Mesoporous Material, 2003, 65, 233-242.
- Eregowda, G. B.; Kalpana, H. N.; Hegbe, R.; Thimmiah, K. N. Ind. J. Chem. Soc.2000, 39B, 24.
- Okafor, C. O. Phosphorus and Sulphur , 1978, 4, 79.
- Newman, M. S.; Perry, C. Y. J. Org. Chem. 1963, 28, 116.
- Ruan, J. W.; Huang, Z. S.; Huang, J. F.; Du, C. J.; Huang, S. L.; Shi, Z.; Fu, L. W.; Gu, L. Q. Chinese Chemical Letters, 2006, 17(9), 06.
- Zaytsey, A. V.; Anderson, R. J.; Bedernjack, A.; Groundwater, P. W.; Huang, Y.; Perry, J. D.; Orenga, S.; Roger-Dalbert, C.; James, A. Org. Biomol. Chem., 2008, 6, 682-692.
- Abe, A.; Yamane, M.; Tomoda, A. Anticancer Drugs 2001, 12 (4), 377-382.
- Gerasimova, T. N.; Kolchina, E. F.; Kargapolova, I. Y.; Fokin, E. P. Russ. J. Org. Chem. 1997, 33, 735–739.
- Jose, J.; Burgess, K. Tetrahedron, 2006, 62, 11021-11037.
- Carr, G.; Tay, W.; Bottriell, H.; Andersen, S. K.; Mauk, A. G.; Andersen, R. J. Org. Lett. 2009, 11, 2996-2999.
- Jeromin, G. E. Tetrahedron Lett. 2001, 42, 1863–1865.
- Thimmaiah, K. N.; Easton, J. B.; Germain, G. S.; Morton, C. L.; Kamath, S.; Buolamwini, J. K..; Houghton, P. J. J. Biol. Chem., 2005, 280(36), 31924-31935.
- Pratt, Y. T.; Drake, N. L. JACS, 1960, 82, 1152-1160.
- Petrov, V.; Sturgeon, B. J. Chem. Soc., 1954, 570-574.
- Furniss, B. S.; Hannaford, A. J.; Rogers, V.; Smith, P. W. G.; Tatchell, A. R. Vogel’s Textbook of Organic Chemistry 4th ed. Longman, England 934-936, 1978.
- Williamson, K. L. Macroscale and Microscale Organic Experiments 3rd ed. Houghton, US, 518, 1999.
- Brian, S. F.; Antony, J. H.; Peter, W. G.; Austin, R. T. Vogel’s Textbook of Organic Chemistry 4th ed. Longman, England, 930-933, 1989.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


