Isolation and Characterization of Selected Impurities Obtained from Nortriptyline
 , Vinayak M. Gaware*, Kiran B.Dhamakand Balu T. Jagtap
, Vinayak M. Gaware*, Kiran B.Dhamakand Balu T. JagtapPRES’s College of Pharmacy (For Women), Chincholi, Tal. Sinnar, Nashik, Maharashtra, India
Corresponding Author E-mail:vinayak.gaware@pravara.in
DOI : http://dx.doi.org/10.13005/ojc/420330
Download this article as:
![]()
This study investigates the degradation behavior of nortriptyline hydrochloride under forced acidic conditions as per ICH Q1A (R2) guidelines. Acid-induced degradation products were isolated using flash chromatography, yielding highly pure fractions. Two major impurities were selected for detailed structural characterization using nuclear magnetic resonance (NMR) spectroscopy and liquid chromatography–mass spectrometry (LC–MS), which revealed significant structural transformations. A robust chromatographic method was developed and validated according to ICH Q2 (R1), demonstrating high precision, accuracy, and repeatability. The study emphasizes the importance of early detection and characterization of degradation impurities to ensure drug quality, stability, and patient safety. This approach provides a practical framework for routine impurity profiling in quality control laboratories and supports the development of stability-indicating methods. The findings offer valuable insight into nortriptyline’s degradation pathways and contribute to predictive strategies for minimizing harmful impurity formation during storage, thereby enhancing overall pharmaceutical safety and efficacy.
KEYWORDS:Forced Degradation; Impurity Profiling; LC–MS Characterization; Nortriptyline Hydrochloride; Stability-Indicating Method
Introduction
Organic impurities, inorganic impurities, and residual solvents are the three primary groups into which pharmaceutical impurities are typically divided. Degradation during storage or the production process might produce organic contaminants. These can include unreacted starting materials, intermediates, or degradation products induced by environmental factors such as heat, light, or moisture, as well as interactions with excipients or packaging components. Volatile organic compounds employed in production or purification are known as residual solvents. If not completely removed, they may remain in trace quantities in the final drug product 1-6. Regulatory authorities such as the International Council for Harmonization (ICH) classify these solvents according to toxicity and specify acceptable limits to ensure patient safety. Controlling impurities is crucial for ensuring product efficacy, stability, and compliance with Good Manufacturing Practices (GMP). Analytical tools such as ICP-MS, GC, and HPLC are employed to detect and quantify impurities. Following ICH guidelines throughout manufacturing and stability testing helps maintain product purity over its entire shelf life 7.
Materials and Methods
UV Spectphotometric Methods for Drug
Preparation Of Standard Solution
Accurately weighed quantity of Nortriptyline 10mg was transferred to 100 ml volumetric flask and volume made up to the mark with distilled water to give a stock solution having strength of 100μg/ml.
From the stock solution of 100μg/ml pipette out 10 ml in 100 ml volumetric flask and volume made up to the mark with a distilled water to give a standard solution of 10μg/ml. absorbance was taken.
Method for Validation
Parameters to be Considered for the Validation of Method are:
Linearity and Range
The linearity response was determined by analyzing 5 independent concentration levels of calibration curve in range of 5-25μg/ml for Nortriptyline. The calibration curve of absorbance vs. respective concentration was plotted and correlation coefficient and regression line equation for Nortriptyline were calculated.
Accuracy
Accuracy may often be expressed as % recovery by the assay of known, added amount of analyte. The recovery experiments were carried out in triplicate by adding analysed samples of the Nortriptyline with three different concentrations of standard at 80%, 100%, and 120%. Absorbance of solution was measured at 279 nm. The amount of Nortriptyline was calculated at each level and % recoveries were computed.
In pure drug proportion was 10 µg/ml i.e. 1 ml; consider as 100% so calculate the 80% and 120% level of recovery and calculated how much standard (pure drug) solution was added into the tablet solution.
Precision
Repeatability: aliquots of 2, 4, and 8 ml of working standard solution (100μg/ml) were transferred to a series of 10ml volumetric flask. The volume was adjusted up to mark with distilled water. The absorbance of above solution was measured three times and % RSD was calculated.
Intraday precision: solutions containing 5, 10, and 15μg/ml of Nortriptyline was prepared and analyzed 3 times on the same day and % RSD was calculated.
Interday precision: solution containing 5, 10, and 15 μg/ml of Nortriptyline was prepared analyzed 3 times on 3 different days and %RSD was calculated.
Robustness
Solution containing 20 μg/ml of Nortriptyline were prepared and analyzed at 2 different temperature and %RSD was calculated.
Limit of Detection and Limit of Quantitation
LOD and the LOQ of the drug were calculated using the following equations as per ICH guidelines.
LOD=3.3 x (SD/slope)
LOQ= 10 x (SD/slope)
Where, SD= the standard deviation of intercept of calibration curves.
Slope= the mean slope of the calibration curve.
Stability of Solvents
The stability for Nortriptyline was found to be in the 25μg/ml for 120min.
Chemical and Material
Nortriptyline was procured as gift sample, Acetonitrile, Methanol, Glacial Acetic Acid, Toluene, Ethyl acetate, Potassium Dihydrogen Phosphate, Sodium Hydroxide, Conc. Hydrochloric acid, and Hydrogen Peroxide was used as HPLC grade from Merck Chemical Ltd.
Sample Preparation and Isolation of Impurities
Forced degradation was carried out under acidic conditions. The acid degradation product was isolated by flash chromatography using a Combi Flash Companion system (Teledyne ISCO) fitted with a Gold 80 column. The mobile phase consisted of methanol and chloroform (1:1, v/v). Detection was primarily at 254 nm with additional monitoring at 287 nm. Fractions were collected in 16 mm × 150 mm tubes (13 mL peak tube volume). For acidic degradation, 200 mg of degradant was adsorbed onto silica gel (60–120 mesh) in a 1:4 ratio (drug: silica gel) and dried under vacuum (<60°C). Column chromatography was performed using a stepwise chloroform–methanol gradient (100:0 to 0:100 v/v). 100 mL of fractions were gathered and condensed at a lower pressure 8.
Characterization IR
A dry sample of the analyte and IR-grade KBr was prepared by drying at 110 °C for at least 2 h to remove moisture properly. The dried sample and KBr were mixed in a ratio of approximately 1–2 mg of sample to 100–200 mg of KBr (0.5–2% w/w) and ground to a very fine, homogeneous powder in agate mortar. The mixture was then pressed into a transparent pellet under a pressure of 8–10 tonnes for 1–2 min. The pellet was handled with gloves to avoid contamination, and the IR spectrum was recorded immediately 9.
Mass Spectroscopy
The mass spectroscopy analysis utilized advanced ionization sources. The instrument featured a high mass resolution of 50,000 Full Sensitivity Resolution (FSR), ensuring precise identification. It offered sub-ppm mass accuracy and a wide mass range of 20 to 3500 m/z, enabling comprehensive analysis of complex samples 10.
NMR
A 500 MHz BrukerAvance III HD spectrometer running Topspin 3.2 software was used to conduct the NMR analysis. The solvent used was DMSO-d6. This setup provides high-resolution nuclear magnetic resonance spectra for detailed structural analysis 11-13.
Results and Discussion
Isolation of Impurities by flash chromatography
Flash chromatography is a valuable purification method that offers rapid separation and high-purity yields, making it particularly useful in organic synthesis, drug discovery, and natural product extraction. The following common solvents are chosen for their best separation and purification efficiency: methanol (density: 0.79 g/mL, elution strength: 0.70), ethyl acetate (density: 0.90 g/mL, elution strength: 0.45), and toluene (density: 0.87 g/mL, elution strength: 0.22). The acid degradant was isolated in four distinct fractions, which were collected in sample vials. These fractions were subsequently subjected to further characterization by LC–MS and NMR (Figure 1 and Figure 2).
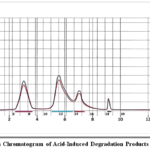 |
Figure 1: Flash Chromatogram of Acid-Induced Degradation Products of Nortriptyline Click here to View Figure |
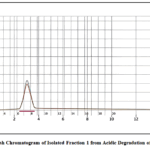 |
Figure 2: Flash Chromatogram of Isolated Fraction 1 from Acidic Degradation of Nortriptyline Click here to View Figure |
Characterization of Impurities IR
The FT-IR spectrum revealed characteristic absorption bands confirming the presence of multiple functional groups. A very intense and broad band at 2500–3300 cm⁻¹ was attributed to the stretching O–H vibration of a carboxylic acid, which overlapped with other absorptions in this region. A strong C=O stretching band was observed at 1700–1725 cm⁻¹, consistent with carboxylic acid or unconjugated ketone functionalities, while a shift to 1735–1750 cm⁻¹ indicated ester carbonyl groups. Conjugation of C=O to an aromatic ring or C=C bond was evident from the lowered stretching frequency in the 1680–1700 cm⁻¹ range. Amide carbonyl absorptions appeared in the 1630–1690 cm⁻¹ range, accompanied by N–H stretching between 3300–3500 cm⁻¹. The aldehyde C=O stretch was detected at 1720–1740 cm⁻¹, with weak C–H overtone bands near 2720–2820 cm⁻¹. Strong absorptions for tertiary amine N-oxides were noted around 1060–1260 cm⁻¹, overlapping with C–O and C–N stretching bands (1250–1020 cm⁻¹). Aromatic C=C stretches were observed at 1600, 1500, and 1475 cm⁻¹, while out-of-plane =C–H bending modes appeared in the 900–700 cm⁻¹ region, useful for determining substitution patterns. Aliphatic C–H stretches were evident between 2950–2850 cm⁻¹. The C–O stretching vibrations of alcohols, ethers, and esters were assigned to strong absorptions in the 1300–1000 cm⁻¹ regions.
LC-Mass Spectrometry
In the LC–MS analysis of the isolated impurity from nortriptyline, a distinct chromatographic peak was observed at a retention time (RT) of 4.57 minutes. The mass spectrometric profile revealed prominent m/z values of 268.17, 274.87, and 302.44, indicating the presence of the protonated molecular ion along with characteristic isotopic and adduct peaks. The observed m/z 268.17 corresponds to the likely [M+H] + species of the impurity, while the minor peaks at m/z 274.87 and 302.44 may be attributed to isotopic variants and possible sodium adducts, respectively. These results confirm the successful isolation of the impurity and provide key molecular mass information essential for its structural elucidation (Figure 3).
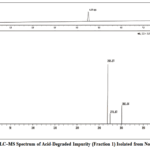 |
Figure 3: LC–MS Spectrum of Acid-Degraded Impurity (Fraction 1) Isolated from Nortriptyline Click here to View Figure |
A Bruker 500 MHz NMR spectrometer (Avance III HD model) with Topspin 3.2 software was used for the NMR analysis. The acid degradant fraction 1 was obtained by evaporating the solvent and dissolving the resulting residue in DMSO. The samples were then analyzed for both proton.
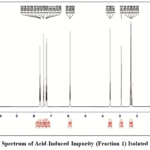 |
Figure 4: ¹H NMR Spectrum of Acid-Induced Impurity (Fraction 1) Isolated from Nortriptyline Click here to View Figure |
The ¹H NMR spectrum (δ, ppm, 400/500 MHz, CDCl₃) shows a cluster of resonances in the aromatic region at δ 7.24–7.68 (multiple signals: 7.246, 7.255, 7.267, 7.281, 7.295, 7.428, 7.432, 7.442, 7.624, 7.638, 7.655, 7.664, 7.682), consistent with one or more substituted phenyl rings. Signals at δ 5.88–5.89 likely arise from vinylic or deshieldedolefinic protons (reported here as 5.882, 5.892). Resonances in the 3.54–3.57 ppm region (3.547, 3.566, and 3.574) are characteristic of methylene or methoxy protons adjacent to oxygen or other electron-withdrawing groups. A singlet/peak at δ ~2.91 (2.913) suggests a benzylic/α-to-heteroatom methine or methylene. Multiple signals between δ 2.29–2.40 (2.293, 2.324, 2.374, 2.394 — with some overlap/repetition) correspond to aliphatic CH₂ groups in slightly different chemical environments, indicating non-equivalent methylene units or conformers. Additional aliphatic resonances observed at δ 1.54 and 2.14–2.35 are consistent with methyl or further methylene groups (Figure 4).
The ¹H NMR spectrum displays multiple resonances across the aliphatic (δ ~1.5–3.6), mid-field (δ ~5.9), and aromatic (δ ~7.24–7.68) regions. In the aliphatic region several closely spaced signals appear between δ 2.29–2.40 (multiple doublets), together with distinct resonances at δ 2.74 (singlet) and δ 3.55–3.57 (doublets), consistent with benzylic or methylene environments and substituted methine/methylene groups. A triplet at δ ~1.54 indicates a terminal methyl group adjacent to a methylene. A pair of resonances at δ ~5.88–5.89 suggests a vinylic or deshieldedoxymethine proton. The aromatic region shows a cluster of signals between δ 7.24 and 7.68 (several closely spaced doublets/multiplets), consistent with a mono- or polysubstituted aromatic ring. Overall, the pattern of doublets in both the aliphatic and aromatic regions is consistent with multiple neighboring proton couplings (J-couplings), while the singlets indicate isolated protons.
Conclusion
The acid degradant of nortriptyline was successfully isolated by flash chromatography into four distinct fractions, with fraction 1 characterized in detail. FT-IR analysis revealed the presence of carboxylic acid, ester, amide, aromatic, and aliphatic functional groups. LC–MS data indicated a principal protonated molecular ion at m/z 268.17, supported by isotopic (m/z 274.87) and sodium adduct (m/z 302.44) peaks, corresponding to the likely molecular mass of the impurity. The ¹H NMR spectrum exhibited resonances in the aromatic, vinylic, and aliphatic regions, consistent with a substituted phenyl ring system bearing electron-withdrawing substituents and multiple methylene/methoxy linkages. Collectively, these results confirm the isolation of a structurally distinct degradation product, providing a basis for further structural elucidation and degradation pathway analysis of nortriptyline under acidic conditions.
Acknowledgement
I acknowledge the support and guidance provided by various individuals and organizations throughout this research journey.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Bhatt, H.,Identification and structural elucidation of stress degradation products in pharmaceutical substances: An advanced LC–MS approach. Journal of Pharmaceutical and Biomedical Analysis, 2022, 231, 115504.
- Kumar, A.,Development and validation of a stability-indicating chromatographic method for impurity profiling of active pharmaceutical ingredients. Journal of Chromatographic Science, 2022, 60(8), 724–734.
- Suryawanshi, R., Forced degradation behavior and LC–MS characterization of acidic impurities formed in psychotropic drug substances. Journal of Applied Pharmaceutical Science, 2021, 11(5), 101–109.
- Zhang, T., Application of high-resolution mass spectrometry in the identification of pharmaceutical degradation products. Analytical and Bioanalytical Chemistry, 2020, 412(12), 2935–2946.
- Shelar, A.,Recent advances in flash chromatography for the isolation of degradation impurities in bulk drugs. Analytical Chemistry Research, 2024, 36, 100527.
- Nouruddin W.A., LC-MS as a stability-indicating method for analysis of hyoscine n-butyl bromide under stress degradation conditions with identification of degradation products, Anal. Acta, 2013, S7, 2–5.
CrossRef - Nageswara R.R., Identification and characterization of stress degradants of lacosamide by LC–MS and ESI-Q-TOF-MS/MS: Development and validation of a stability indicating RP-HPLC method, Pharm. Biomed. Anal., 2014, 95, 256–264.
CrossRef - Chunyong W., Degradation kinetics study of cabozantinib by a novel stability-indicating LC method and identification of its major degradation products by LC/TOF-MS and LC–MS/MS, Pharm. Biomed. Anal., 2014, 98, 356–363.
CrossRef - Carstensen J.T., Rhodes C.T., Drug Stability: Principles and Practices, 3rd Edn., Marcel Dekker, New York, 2000, 146–153.
- Mishra J., Nayak S.K., Development, validation and stability study of UV spectrophotometric method for determination of repaglinide in bulk and pharmaceutical dosage forms, Innov. Appl. Pharm. Sci., 2016, 3, 10–16.
- Kumar D.G., Flash chromatography and its different dissolvable frameworks: A review, Chem., 2016, 55–67.
- Indumathi K.V., Isolation and characterization of forced degradative products of an antipsychotic drug levosulpiride by spectroscopic techniques, Chem. Pharm. Res., 2016,8, 1100–1106.
- Pasha S., Khanam S., Isolation and characterization of chemical constituents of Asparagus racemosus as markers, J. Res. Dev. Pharm. Life Sci., 2016, 5, 2255–2263.
Accepted on: 02 Mar 2026
Second Review by: Dr. Astha Jaiswal
Final Approval by: Dr. Murat Hatipoğlu







ISSN Online: 2231-5039

![]()




{kind=link}