Impact of a Novel Dehydroepiandrosterone Derivative on the Metabolism of RWPE-1 and LNCaP Cell Lines
 , Karla Mejía1, Ivonne Heuze1, Miguel Morales-Pacheco2and Mauricio Rodríguez-Dorantes2
, Karla Mejía1, Ivonne Heuze1, Miguel Morales-Pacheco2and Mauricio Rodríguez-Dorantes21Departamento de Sistemas Biológicos y de Producción Agrícola y Animal, Universidad Autónoma Metropolitana-Xochimilco. Calzada del Hueso 1100, Colonia Villa Quietud, Coyoacán, Ciudad de México.
2Laboratorio de Oncogenómica, Instituto Nacional de Medicina Genómica. Periférico Sur, Arenal Tepepan, Tlalpan, Ciudad de México, CDMX.
Corresponding Author E-mail:marisa@correo.xoc.uam.mx
DOI : http://dx.doi.org/10.13005/ojc/420336
Download this article as:
![]()
The distinctiveness of this article lies in its groundbreaking findings on the activity of the dehydroepiandrosterone derivative (12). In both normal (RWPE-1) and tumorigenic (LNCaP) prostate cells, this compound significantly reduces cell viability and increases apoptosis when cells are stimulated with testosterone. In stark contrast, treatment with dihydrotestosterone plus 12 does not diminish viability or apoptosis, thereby demonstrating its mechanism of action. Furthermore, when reference compounds for 5α-reductase (SRD5A1/2) inhibition, such as finasteride and dutasteride, are used, the behavior of these cells diverges sharply from that observed with derivative 12 under stimulation by testosterone and dihydrotestosterone. In this study, we demonstrated that finasteride inhibited testosterone-stimulated growth of RWPE-1 cells, whereas dutasteride was even more effective. However, both drugs also reduced RWPE-1 cell proliferation and increased apoptosis in the presence of dihydrotestosterone, suggesting that they may interact with the androgen receptor (AR). Furthermore, steroid 12 was more effective than finasteride at blocking testosterone-stimulated growth; however, it did not affect dihydrotestosterone-stimulated growth in RWPE-1 cells. The results also showed that treating testosterone-stimulated LNCaP cells with Finasteride, dutasteride, or the novel steroid 12 reduced cell viability and increased apoptosis. Additionally, treating dihydrotestosterone-stimulated LNCaP cells with 1 nMdutasteride reduced their viability and increased apoptosis, as observed in RWPE-1 cells. However, dihydrotestosterone-stimulated LNCaP cells responded differently to finasteridethan RWPE-1 cells; specifically, LNCaP cell viability did not decrease under these conditions. In conclusion, RWPE-1 cells are suitable for evaluating the pharmacological activity of novel drugs on LNCaP tumor cell viability. In this model, finasteride and dutasteride reduce cell viability and increase apoptosis when cells are stimulated with testosterone or dihydrotestosterone. However, the novel derivative 12 reduces viability and induces apoptosis only in testosterone-stimulated normal and tumor cells.
KEYWORDS:Castration-Resistant Prostate Cancer; Finasteride; Cellcomparisonmodels; Dehydroepiandrosteronederivative; Dutasteride; LNCa Pcellproliferation; RWPE-1 cell line
Introduction
Despite ongoing efforts by pharmaceutical companies to improve prostate cancer treatments, the disease still leads to death in at least 16.5% of patients with metastatic cancer, which affects roughly 21% of those diagnosed. Global Cancer Research highlights these concerns.1 This grim statistic is primarily due to prostate tumors’ ability to produce steroids that significantly influence their growth and metabolism.2
Male hormonesstimulate the growth of androgen-dependent tissues, thereby facilitating the development of secondary sexual characteristics in males. Testosterone (T), predominantly produced in the testes, is released into the bloodstream and subsequently absorbed by the prostate gland .3 Furthermore, the prostate gland can synthesize T from circulating steroids originating from the adrenal cortex. These steroids include dehydroepiandrosterone, androstenedione, 11β-hydroxyandrostenedione (11OHA4), and 11β-hydroxytestosterone.4,5
Testosterone is converted to dihydrotestosterone (DHT) by the enzyme 5α-reductase (SRD5A) in the cytoplasm of prostate cells.3 Two isoforms of this enzyme, type 1 (SRD5A1) and type 2 (SRD5A2), are present in this gland.3 In prostatic tumors, type 1 is more prominently expressed than type 2.6 Conversely, in healthy prostate tissue, SRD5A2 is the primary isoform.6 DHT is a more potent metabolite than T in stimulating protein synthesis and, consequently, the growth of androgen-dependent tissues.7
Once synthesized in the cytoplasm, dihydrotestosterone (DHT) binds to the androgen receptor (AR). In its inactive state, the AR is associated with heat-shock chaperone proteins. Upon DHT binding to the AR, the complex dissociates from these chaperones, undergoes a conformational change, and is transported to the nucleus as a dimer. Dimerization occurs through interactions between the N-terminal transactivation domain (TAD) of one monomer and the C-terminal ligand-binding domain (LBD) of the other. DHT has a higher affinity for the AR than testosterone (T); however, T can also enter the nucleus via the AR.
AR comprises three primary functional domains: the N-terminal domain (NTD), the DNA-binding domain (DBD), and the C-terminal ligand-binding domain (LBD). The LBD connects to the DBD via a flexible hinge region formed by residues 623–665. Within the AR are two functional segments: AF1 (amino acids 103-372 and 362-538), located within the NTD and constitutively active, and AF2, located within the LBD and dependent on ligand binding for activation7,8 AR activation occurs when the NTD interacts with the LBD in the presence of bound androgen.7,8 In addition, when the DBD binds the promoter and enhancer regions of specific androgen-regulated genes, transcription of genes linked to the NTD is stimulated. This process recruits coregulators to the AF1 domain within the NTD. As a result, some AR target-linked genes are transcribed.9
To investigate mechanisms previously described in pathologies such as prostate cancer, researchers often use various cancer cell lines. However, a significant gap in these studies is the lack of comparisons with cell lines derived from the exact embryological origin that show no signs of pathological alterations. Consequently, the goal of this research was to determine the effect of a novel dehydroepiandrosterone derivative, previously identified as an inhibitor of SRD5A (10), and to establish a comparative cell line model that facilitates interpretation of results from cell proliferation and apoptosis assays in the presence of dutasteride and finasteride.
Materials
Testosterone (T) was obtained from Ehrenstorfer (LGC, Mexico). Dihydrotestosterone (DHT) and androstenedione (4-androstene-3,17-dione, 4-dione) were acquired from Steraloids (Wilton, NH, USA). The LNCaP and RWPE-1 cells were sourced from the American Type Culture Collection (ATCC, Manassas, VA). Sigma supplied Finasteride (FIN) and Dutasteride (USP Reference Standard). Steroid 12 was previously synthesized and characterized by our research group as an inhibitor of human prostate SRD5A1, as documented in Bratoeff et al.10 (Figure 1). Dr. Eugene Bratoeff generously donated the derivatives to our lab for further study of their potential to reduce LNCaP cell viability.
 |
Figure 1: Structures of Finasteride, Dutasteride, Dehydroepiandrosterone, and the previously synthesized Dehydroepiandrosterone Derivative 12. Click here to View Figure |
Experimental
Determining the optimal concentration of androgens for stimulating proliferation in RWPE-1 and LNCaP cells.
RWPE-1 cells (passages 10–20) were obtained from ATCC and cultured in a Petri dish containing Keratinocyte-SFM medium (GIBCO, CientíficaSenna, CDMX). The medium contained 0.05 mg/mL bovine pituitary extract and 5 ng/mL epidermal growth factor (GIBCO), along with an antibiotic/antimycotic solution (Cytiva, CientíficaSenna, CDMX). RWPE-1 cells were incubated for 7 days. The culture medium was replaced every 3 days, and the cells were maintained at 37 °C in a humidified atmosphere of 5% CO₂ and 95% air in a VWR Symphony 1.4 A incubator (VWR, Mexico City). Once the cells reached confluence, they were washed, trypsinized, and counted. LNCaP cell cultures were obtained as previously described.11
RWPE-1 and LNCaP cells were seeded separately at 12,000 cells per well in distinct 96-well plates. Phenol-free RPMI-1640 medium (Roswell Park Memorial Institute), without antibiotics, bovine pituitary extract, or epidermal growth factor, was then added. For LNCaP cells, the medium was supplemented with 10% activated charcoal-washed fetal bovine serum (GIBCO) and 2 mM glutamine. Both cell types were allowed to adhere for 24 hours. Afterward, the RWPE-1 and LNCaP cell monolayers were washed with phosphate-buffered saline (PBS). Next, different concentrations of T, DHT, or 4-dione (ranging from 10-10 to 10-4 M) were added to each well, dissolved in sterile 0.5% dimethyl sulfoxide(DMSO). All assays were performed in quadruplicate technical replicates across three biological replicates.
The cells were incubated for 24 hours at 37 °C in a humidified atmosphere of 5% CO₂/95% air in a VWR incubator. RWPE-1 and LNCaP cell metabolism was assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method.11 After incubation with 12 mM MTT, formazan crystals formed and were dissolved in 100 μL of DMSO. We determined cell viability by measuring absorbance at 570 nm using a Biotek Epoch Microplate Spectrophotometer (Thermo Fisher Scientific, USA).
Evaluation of the Effects of Finasteride, Dutasteride, and Dehydroepiandrosterone Derivative 12 on the Viability of RWPE-1 Cells
After a 24-hour incubation under specified culture conditions, the RWPE-1 cell layers were washed with phosphate-buffered saline (PBS). Subsequently, FIN, dutasteride, or derivative 12, at concentrations ranging from 10-10 to 10-4 M, was added separately to each well’s cell layer. These compounds were dissolved in sterile 0.5% dimethyl sulfoxide (DMSO). Along with these treatments, 1 nM T or 100 pM DHT, also dissolved in 0.5% DMSO, were included in the experimental setup. All assays were performed in quadruplicate technical replicates across three biological replicates.
The vehicle (DMSO) was added separately to wells containing the RWPE-1 cell layer and served as a viability control. All assays were performed in quadruplicate technical replicates across three biological replicates.
The plate was incubated for 24 hours under the conditions described in Section 3.1. LNCaP cell proliferation was assessed using the MTT method outlined above.
Assessment of Finasteride, Dutasteride, and Derivative 12 on Proliferation of LNCaPCells
LNCaP cell monolayers were washed with PBS. Subsequently, various concentrations of FIN, dutasteride, or derivative 12 (10-10 to 10-4 M), each dissolved in 0.5% DMSO, were added separately to each well. Additionally, 1 nM T or 100 pM DHT, also dissolved in 0.5% DMSO, were added to the same well. Cell incubation conditions and growth assessment were similar to those described in section 3.1. All assays were performed in quadruplicate technical replicates across three biological replicates.
Determination of the Apoptotic Percentage in Cell Cultures
RWPE-1 and LNCaP cells were maintained in the culture media described in section 3.1. Cells (12 X 103) were seeded in 96-well plates, as outlined in section 3.1, and treated with DMSO, FIN, dutasteride, or derivative 12 at concentrations ranging from 10⁻¹⁰ to 10⁻⁴ M, with each treatment applied separately to the cell layer in each well. Additionally, 1 nM T or 100 pM DHT was included, as explained previously. All assays were performed with four technical replicates across three biological replicates. Cell incubation conditions and viability assessment were similar to those described in section 3.1.
After 24 h of incubation, the cells were washed with PBS, and 100 µL of 4% paraformaldehyde in PBS (pH 7.4) was added for 5 to 10 minutes, as previously reported.12 The cells were then transferred to tubes and centrifuged at 1300 rpm for 3 minutes at 4 °C, after which the supernatant was removed. The cells were washed three times with cold PBS and resuspended in a solution containing 1 mMCaCl₂, 0.1 mMdithiothreitol, and 0.1 nM fluorescein-12-2′-deoxyuridine-5′-triphosphate (F-12-dUTP, Merck, CDMX). The tubes were incubated on an ice bed for 10 minutes. After this time, deoxynucleotidetransferase (TdT, Promega, CDMX) was added to the tubes, and the tubes were incubated at 37 °C for 1 hour. The reaction was stopped by adding 2 µL of 0.5 M EDTA. The cells were then washed three times with PBS and immediately examined by phase-contrast and fluorescence microscopy to detect apoptotic cells. Positive cells emit a fluorescent signal, whereas viable cells do not. Two independent observers, blinded to the treatment of each cell group, carried out the microscopic examination. Cells were analyzed in a randomly selected field at 400 x magnification, and 500 nuclei were counted to determine the number of positive cells. The mean immunoreactivity was calculated as the average number of positively stained cells.
Statistical methods
The experimental data were analyzed using a one-way ANOVA in JMP 18, followed by either Dunnett’s or Tukey’s post hoc tests. Dunnett’s test allowed for pairwise comparisons against a defined control group, while Tukey’s test identified which specific group means differed significantly after the ANOVA indicated an overall significant difference. Additional statistical details can be found in the supplementary material.
In the statistical analysis, the viability data from T, DHT, or 4-dione were treated as independent variables. In contrast, the results obtained from finasteride (FIN), dutasteride, or compound 12 were treated as dependent variables. This analysis revealed interactions between the pharmacological agents and hormonal substrates, with statistical significance set at p < 0.05. Further details regarding these tests can be found in the supplementary material.
Results and Discussion
Treatment of RWPE-1 and LNCaP Cells with Androgens
Treatment with 100 pM DHT produced a statistically significant increase in the growth of normal prostate epithelial cells (RWPE-1) within the first 24 hours (P< 0.05), as shown in Figure 2. This result aligns with data reported by Bello et al.13, who observed enhanced proliferation of RWPE-1 cells after exposure to the androgen mibolerone. In contrast, treatment with 1 nM 4-dione significantly suppressed RWPE-1 cell proliferation (P< 0.05), Figure 2.
RWPE-1 cells lack invasive potential, as they do not form clusters or tumors, as noted previously .13 These cells express the androgen receptor in the presence of androgens and encode several enzymes involved in cholesterol-derived steroidogenesis. Major enzymes include CYP11A1, CYP17A1, HSD3β2, and HSD17β3 (AKR1C3). Notably, RWPE-1 cells produce more DHT than T from cholesterol.14
Data also indicated that LNCaP cells responded more strongly to androgenic stimuli, specifically T and DHT, than RWPE-1 cells. This heightened response was associated with a significantly faster cell proliferation rate (Figure 2). Additionally, administering 4-dione significantly promoted LNCaP cell growth at 100 pM and 1 nM, with a p-value < 0.05 (Figure 2). Notably, despite their epithelial origin, LNCaP cells tend to grow in clusters, exhibit significant invasiveness, and form tumors.15 Furthermore, these cells overexpress the AR, even in the absence of androgens in their environment. They also express steroidogenic enzymes, including CYP17, HSD3β2, and AKR1C3, with AKR1C3 being specifically overexpressed in these cells.16,19 However, unlike RWPE-1 cells, they do not encode CYP11A1.
Due to LNCaP cells’ ability to produce more DHT than T and their AR overexpression, the activated receptor can bind chromatin effectively at a ligand concentration that is 100-fold lower. This phenomenon explains the observed increase in LNCaP cell proliferation, as measured at a DHT concentration of 100 pM in the experiment.
The adrenal androgen 4-dione is a circulating androgen that contributes to DHT synthesis in androgen-dependent tissues via the enzyme AKR1C3. AKR1C3 is often overexpressed in prostate tumors, and increased androgen production can promote tumor growth, particularly in the early stages of the disease. In contrast, the low activity of AKR1C3 in RWPE-1 cells, together with their loss of androgen receptor (AR) expression during culture, may explain why these cells do not respond to 4-dione.
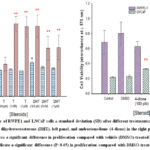 |
Figure 2: Cell viability of RWPE1 and LNCaP cells ± standard deviation (SD) after different treatments: testosterone (T) or dihydrotestosterone (DHT), left panel, and androstenedione (4-dione) in the right panel. |
The asterisk (*) indicates a significant difference in proliferation compared with vehicle (DMSO)-treated RWPE-1 cells. Two asterisks (**) indicate a significant difference (P<0.05) in proliferation compared with DMSO-treated LNCaP cells.
Effect of Steroidal Combination on the Viability of RWPE-1 and LNCaP Cell Lines
Treatment of RWPE-1 cells with both androgens and finasteride reduced their growth. Additionally, treatment with different concentrations of compound 12 (Figure 3) significantly decreased cell growth (P < 0.05). This indicates that compound 12 is more potent than finasteride at inhibiting RWPE-1 cell proliferation. Figure 3 shows that higher concentrations of the inhibitor (12) result in a more pronounced suppression of cell growth.
Finasteride is a type 2 inhibitor of SRD5A222 that reduces DHT production. DHT is a potent androgen that promotes cell proliferation. However, combining DHT with finasteride reduced RWPE-1 cell viability (Figure 3). This unexpected finding suggests that finasteride may interact with the AR because of its structural similarity to DHT.22
Prostate cancer cell models show variable sensitivity to the drugs dutasteride and finasteride when stimulated with DHT, with LNCaP cells the most sensitive. Additionally, when the AR is transfected into drug-sensitive cells—regardless of AR genotype—treatment with both finasteride and dutasteride inhibits AR signaling, with dutasteride showing greater potency than finasteride. In these contexts, disruption of AR function is associated with reduced cell growth.24
The results also showed that combining dutasteride (1 nM) with T or DHT significantly reduced RWPE-1 cell proliferation (P < 0.05). Furthermore, when T was combined with steroid 12 (Figure 3), the same result as in the previous experiment was observed. However, in this case, the effect depended on the concentration used (P< 0.05; Figure 3). Dutasteride has previously been identified as an inhibitor of SRD5A types 1 and 2, rather than as an AR antagonist.24 Data showing that DHT combined with dutasteride (at a concentration of 1 nM) reduced the viability of RWPE-1 cells suggests that the AR may also play a role when dutasteride is present. Overall, the data indicate that derivative 12 exhibits a more selective mechanism of action than finasteride or dutasteride, as it did not affect RWPE-1 cell viability in the presence of DHT.
The crystal structure of the AR has been studied previously.8 Although the mechanism of agonist binding to the AR is well understood, the structural basis of AR antagonism remains unclear8,25
A key region of the AR that changes shape upon antagonist binding is AF-2. This region, located in the C-terminal ligand-binding domain (LBD) of AR, plays a crucial role in regulating interactions among the amino- and carboxy-terminal domains and with coregulators (26-28). These interactions influence the stability of the AR and its DNA-binding affinity.26-30
Modifying an estrogen receptor antagonist that binds the AF-2 motif enhances its interaction with the androgen receptor’s AF-2 domain, thereby reducing transcriptional activity (8, 29). This insight has prompted researchers to develop more effective AF-2 antagonists. Virtual docking analysis has identified several molecules that bind the AF-2 domain.30
In Figure 4, LNCaP cell viability increased with 1 nM T and 100 pM DHT compared with vehicle-treated cells. Adding 25 nMfinasteride (FIN), 1 nMdutasteride, or steroid 12 in combination with T produced a statistically significant decrease in cell viability (P< 0.05). Furthermore, combining DHT with finasteride or steroid 12 did not produce any observable effects. Conversely, combining DHT with dutasteride significantly reduced LNCaP cell proliferation (P < 0.05). These findings are consistent with Chhipa et al.23
Compound 12 inhibits SRD5A1 enzyme activity (10). Data from the experiment reported here showed that 12is more potent than finasteride at reducing RWPE-1 cell proliferation. Finasteride specifically targets SRD5A2, whereas dutasteride is a dual inhibitor of SRD5A1 and SRD5A2. Dutasteride was more potent than both finasteride and compound 12 at inhibiting RWPE-1 cell growth. This indicates that both SRD5A1 and SRD5A2 isotypes are present in RWPE-1 cells.
LNCaP cells express both SRD5A1 and SRD5A2 isotypes, with higher SRD5A1 expression.22,23 The proliferation data from this study indicated that finasteride, dutasteride, and steroid 12 reduced the viability of LNCaP cells when stimulated with T. Additionally, the viability of these cells was decreased when treated with a combination of DHT and dutasteride (1 nM), similar to the effects observed in RWPE-1 cells.
The growth of LNCaP cells in the presence of both FIN and DHT followed a distinct pattern compared with RWPE-1 cells; specifically, LNCaP cell viability did not decrease, whereas RWPE-1 cell viability decreased.
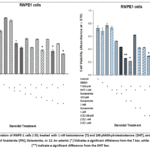 |
Figure 3: Proliferation of RWPE-1 cells ± SD, treated with 1 nM testosterone (T) and 100 pMdihydrotestosterone (DHT), and with different concentrations of finasteride (FIN), Dutasteride, or 12. |
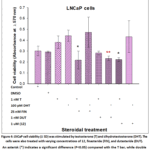 |
Figure 4: LNCaP cell viability (± SD) was stimulated by testosterone (T) and dihydrotestosterone (DHT). The cells were also treated with varying concentrations of 12, finasteride (FIN), and dutasteride (DUT). |
Effect of Steroidal Combination on the Apoptosis of RWPE-1 and LNCaP Cell Lines
RWPE-1 cells treated with DMSO, T, or DHT showed only a few apoptotic cells (see Table 1). In contrast, FIN, when combined with T or DHT, produced a higher percentage of apoptotic cells across the tested doses. Furthermore, derivative 12, in combination with T, induced significant, dose-dependent cell death (P<0.05) compared with T alone (see Table 1). These findings align with the results of the viability studies.
Treatment with dutasteride or the steroid 12, combined with T, increased apoptosis in RWPE-1 cells (P<0.05), as expected (see Table 1). Furthermore, the combination of dutasteride and DHT at 1 nM also increased cell death, indicating an interaction between dutasteride and the AR. Notably, dutasteride treatment in RWPE-1 cells induced greater apoptotic activity than FIN when combined with androgens. Previous studies have demonstrated that dutasteride is more effective than FIN at inhibiting cell growth in RWPE-1 cells,32 findings that align with those reported here.
When T-stimulated LNCaP cells were treated with FIN, dutasteride, and steroid 12, the number of apoptotic cells increased compared with cells stimulated with T alone, owing to the inhibitory effects of these steroids on the SRD5A1/2 isoenzymes. Results with FIN are consistent with studies by Golbano et al.33 However, consistent with the viability experiment reported here, DHT-stimulated LNCaP cells treated with FIN did not show apoptosis. In contrast, dutasteride-treated DHT-stimulated LNCaP cells exhibited increased apoptosis.
Table 1: Percentage ± standard deviation of DHT-stimulated normal RWPE1 and tumorigenic LNCaP apoptotic cells treated with different steroids.
| CelType | DMSO (0.5%) | DHT
(100 pM) |
DHT+FIN
(1 nM) |
DHT+FIN
(25 nM) |
DHT+DUT (100 pM) | DHT+DUT
(1 nM) |
DHT+DUT
(1 uM) |
DHT +12
(100 pM) |
DHT +12
(1 nM) |
DHT+12
(1 uM) |
|
RWPE-1 |
0.4±0.08 | 1.4±.0.1 | 10±0.7 | 22±1.0 | 20±1.2 | 40±0.3 | 15±0.5 | 1.0±0.006 | 1.0±0.5 | 1.0±0.01 |
| LNCaP | 0.25±0.04 | 0.33±0.034 | 1.2±0.05 | 1.1±0.7 | 25±1.5 | 60±10.3 | 55±3.0 | 1.2±0.9 | 0.2±0.001 |
0.5±0.002 |
**DMSO**: Dimethyl Sulfoxide
– **T**: Testosterone
– **DHT**: Dihydrotestosterone
– **FIN**: Finasteride
– **DUT**: Dutasteride
– **DHEA** Dehydroepiandrosterone Derivative “12”
Conclusion
This experiment demonstrated that FIN inhibited the development of T-stimulated RWPE-1 cells. However, RWPE-1 cells showed a stronger response to dutasteride than to FIN, resulting in decreased cell growth. Furthermore, both drugs significantly reduced RWPE-1 proliferation under DHT stimulation, suggesting that FIN and dutasteride may interact with the AR. Both medications are commercially available for the treatment of benign prostatic hyperplasia.
Furthermore, the new dehydroepiandrosterone derivative 12 was more effective than FIN at inhibiting T-stimulated RWPE-1 cell growth. However, it failed to reduce DHT-stimulated RWPE-1 cell growth, suggesting that this compound did not interact with the AR.
The results also displayed that T-stimulated LNCaP cells treated with FIN, dutasteride, or the novel steroid 12 exhibited reduced viability. Additionally, the viability of DHT-stimulated LNCaP cells decreased upon dutasteride treatment (1 nM), consistent with the effects observed in RWPE-1 cells. However, the growth of DHT-stimulated LNCaP cells in the presence of FIN differed from that of RWPE-1 cells; specifically, LNCaP cells did not show decreased viability. This finding suggests that a FIN could interact with the AR in both LNCaP and RWPE-1 cells.
The findings suggest that RWPE-1 cells could serve as a useful paradigm for testing the in vitro effects of new drugs designed to reduce the viability of tumoralLNCaP cells and to induce apoptosis.
Acknowledgement
This study was conducted with funds approved by the Universidad AutónomaMetropolitana-Xochimilco for the project “Mechanism of Action of Steroid Hormones in Different Tissues.” The project was approved by the Publication Divisional Council on July 24, 2024, during its 14th session of 2024, with agreement 14/24.3.8.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
The data for this experiment are available in the supplementary material file and the repositories of the Hormone Laboratory of Universidad AutónomaMetropolitana-Xochimilco
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Authors Contributions
All authors contributed equally to the conception, development, and preparation of this work. All authors reviewed and approved the final manuscript and consented to its submission for publication.
References
- Global Cancer Research highlights these concerns. Recovered from: https://gco.iarc.fr/:Global: Global: Global: Global Cancer Observatory, World Health Organization, International Agency for Research on Cancer.
- Yepuru, M.; Wu, Z.; Kulkarni, A.; Yin, F.; Barrett, C. M.; Kim, J.; Steiner, M. S.; Miller, D. D.; Dalton, J. T.; Narayanan, R. Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res 2013, 19 (20), 5613–5625. DOI: 10.1158/1078-0432.ccr-13-1151.
CrossRef - Russell, D. W.; Wilson, J. D. Steroid 5α-Reductase: Two Genes/Two Enzymes. Annual Review of Biochemistry 1994, 63 (1), 25-61. DOI: 10.1146/annurev.bi.63.070194.000325.
CrossRef - Capper, C. P.; Rae, J. M.; Auchus, R. J. The Metabolism, Analysis, and Targeting of Steroid Hormones in Breast and Prostate Cancer. Horm Cancer 2016, 7 (3), 149-164. DOI: 10.1007/s12672-016-0259-0.
CrossRef - Turcu, A. F.; Rege, J.; Auchus, R. J.; Rainey, W. E. 11-Oxygenated androgens in health and disease. Nat Rev Endocrinol 2020, 16 (5), 284-296. DOI: 10.1038/s41574-020-0336-x.
CrossRef - Thomas, L. N.; Lazier, C. B.; Gupta, R.; Norman, R. W.; Troyer, D. A.; O’Brien, S. P.; Rittmaster, R. S. Differential alterations in 5α-reductase type 1 and type 2 levels during development and progression of prostate cancer. The Prostate 2005, 63 (3), 231-239. DOI: 10.1002/pros. 20188.
CrossRef - Obst, J. K.; Tien, A. H.; Setiawan, J. C.; Deneault, L. F.; Sadar, M. D. Inhibitors of the transactivation domain of androgen receptor as a therapy for prostate cancer. Steroids 2024, 210, 109482. DOI: 10.1016/j.steroids.2024.109482.
CrossRef - Tan, M. H.; Li, J.; Xu, H. E.; Melcher, K.; Yong, E. L. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol Sin 2015, 36 (1), 3-23. DOI: 10.1038/aps. 2014.18.
CrossRef - Jenster, G.; van der Korput, H. A.; Trapman, J.; Brinkmann, A. O. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem 1995, 270 (13), 7341-7346. DOI: 10.1074/jbc. 270.13.7341.
CrossRef - Bratoeff, E.; Sánchez, A.; Arellano, Y.; Heuze, Y.; Soriano, J.; Cabeza, M. In vivo and in vitro effect of androstene derivatives as 5α-reductase type 1 enzyme inhibitors. J Enzyme Inhib Med Chem 2013, 28 (6), 1247-1254. DOI: 10.3109/14756366.2012.729827.
CrossRef - Cabeza M, Menes L. A, Fuentes E, Bahena I, Heuze Y. Novel Type 1 5α-Reductase Inhibitors with Antiproliferative Potential on LnCaP Cells. Orient J Chem. 2023;39(3),523-532 DOI:10.13005/ojc/390301
CrossRef - Piqueras, B.; Autran, B.; Debre, P.; Gorochov, G. Detection of Apoptosis at the Single-Cell Level by Direct Incorporation of Fluorescein-dUTP in DNA Strand Breaks. BioTechniques 1996, 20 (4), 634-640. DOI: 10.2144/19962004634.
- Bello, D.; Webber, M. M.; Kleinman, H. K.; Wartinger, D. D.; Rhim, J. S. Androgen-responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus. Carcinogenesis 1997, 18 (6), 1215-1223. DOI: 10.1093/carcin/18.6.1215.
CrossRef - Bennett, N. C.; Hooper, J. D.; Lambie, D.; Lee, C. S.; Yang, T.; Vesey, D. A.; Samaratunga, H.; Johnson, D. W.; Gobe, G. C. Evidence for Steroidogenic Potential in Human Prostate Cell Lines and Tissues. The American Journal of Pathology 2012, 181 (3), 1078-1087. DOI 10.1016/j.ajpath.2012.06.009.
CrossRef - Igawa, T.; Lin, F.-F.; Lee, M.-S.; Karan, D.; Batra, S. K.; Lin, M.-F. Establishment and characterization of an androgen-independent human prostate cancer LNCaP cell model. The Prostate 2002, 50 (4), 222-235. DOI: doi.org/10.1002/pros.10054.
CrossRef - Urbanucci, A.; Sahu, B.; Seppälä, J.; Larjo, A.; Latonen, L. M.; Waltering, K. K.; Tammela, T. L. J.; Vessella, R. L.; Lähdesmäki, H.; Jänne, O. A. Overexpression of androgen receptor enhances the binding of the receptor to the chromatin in prostate cancer. Oncogene 2012, 31 (17), 2153-2163.DOI: 10.1038/onc.2011.401.
CrossRef - Kobayashi, T.; Inoue, T.; Kamba, T.; Ogawa, O. Experimental evidence of persistent androgen-receptor-dependency in castration-resistant prostate cancer. Int J Mol Sci 2013, 14 (8), 15615-15635. DOI: 10.3390/ijms140815615.
CrossRef - Byrns, M. C.; Jin, Y.; Penning, T. M. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): overview and structural insights. J Steroid Biochem Mol Biol 2011, 125 (1-2), 95-104. DOI: 10.1016/j.jsbmb.2010.11.004.
CrossRef - Hertzog, J. R.; Zhang, Z.; Bignan, G.; Connolly, P. J.; Heindl, J. E.; Janetopoulos, C. J.; Rupnow, B. A.; McDevitt, T. M. AKR1C3 mediates pan-AR antagonist resistance in castration-resistant prostate cancer. Prostate 2020, 80 (14), 1223-1232. DOI: 10.1002/pros. 24049.
CrossRef - Kukkonen, K.; Autio-Kimura, B.; Rauhala, H.; Kesseli, J.; Nykter, M.; Latonen, L.; Visakorpi, T. Nonmalignant AR-positive prostate epithelial cells and cancer cells respond differently to androgen. Endocr Relat Cancer 2022, 29 (12), 717-733. DOI: 10.1530/erc-22-0108.
CrossRef - Bull, H. G.; Garcia-Calvo, M.; Andersson, S.; Baginsky, W. F.; Chan, H. K.; Ellsworth, D. E.; Miller, R. R.; Stearns, R. A.; Bakshi, R. K.; Rasmusson, G. H.; et al. Mechanism-based inhibition of human steroid 5α-reductase by finasteride: Enzyme-catalyzed formation of NADP-dihydrofinasteride, a potent bisubstrate analog inhibitor. Journal of the American Chemical Society 1996, 118 (10), 2359-2365. DOI: 10.1021/ja953069t
CrossRef - Wu, Y.; Chhipa, R. R.; Zhang, H.; Ip, C. The antiandrogenic effect of finasteride against a mutant androgen receptor. Cancer Biol Ther 2011, 11 (10), 902-909. DOI: 10.4161/cbt. 11.10.15187.
CrossRef - Chhipa, R. R.; Halim, D.; Cheng, J.; Zhang, H. Y.; Mohler, J. L.; Ip, C.; Wu, Y. The direct inhibitory effect of dutasteride or finasteride on androgen receptor activity is cell line-specific. The Prostate 2013, 73 (14), 1483-1494. DOI: 10.1002/pros. 22696
CrossRef - Lazier, C. B.; Thomas, L. N.; Douglas, R. C.; Vessey, J. P.; Rittmaster, R. S. Dutasteride, the dual 5α-reductase inhibitor, inhibits androgen action and promotes cell death in the LNCaP prostate cancer cell line. The Prostate 2004, 58 (2), 130-144: DOI: 10.1002/pros. 10340.
CrossRef - Pereira de Jésus-Tran, K.; Côté, P.-L.; Cantin, L.; Blanchet, J.; Labrie, F.; Breton, R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Science 2006, 15 (5), 987-999. DOI: 0.1110/ps. 051905906.
CrossRef - He, B.; Wilson, E. M. The NH(2)-terminal and carboxyl-terminal interaction in the human androgen receptor. Mol Genet Metab 2002, 75 (4), 293-298. DOI: 10.1016/s1096-7192(02)00009-4.
CrossRef - Hsu, C. L.; Chen, Y. L.; Yeh, S.; Ting, H. J.; Hu, Y. C.; Lin, H.; Wang, X.; Chang, C. The use of the phage display technique for the isolation of androgen receptor-interacting peptides with (F/W)XXL(F/W) and FXXLY new signature motifs. J Biol Chem 2003, 278 (26), 23691-23698. DOI: 10.1074/jbc.M211908200.
CrossRef - He, B.; Lee, L. W.; Minges, J. T.; Wilson, E. M. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. J Biol Chem 2002, 277 (28), 25631-25639. DOI: 10.1074/jbc.M202809200.
CrossRef - He, B.; Minges, J. T.; Lee, L. W.; Wilson, E. M. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J Biol Chem 2002, 277 (12), 10226-10235. DOI: 10.1074/jbc.M111975200.
CrossRef - Gunther, J. R.; Parent, A. A.; Katzenellenbogen, J. A. Alternative inhibition of androgen receptor signaling: peptidomimetic pyrimidines as direct androgen receptor/coactivator disruptors. ACS Chem Biol 2009, 4 (6), 435-440. DOI: 10.1021/cb900043e.
CrossRef - Axerio-Cilies, P.; Lack, N. A.; Nayana, M. R.; Chan, K. H.; Yeung, A.; Leblanc, E.; Guns, E. S.; Rennie, P. S.; Cherkasov, A. Inhibitors of androgen receptor activation function-2 (AF2) site identified through virtual screening. J Med Chem 2011, 54 (18), 6197-6205. DOI: 10.1021/jm200532b.
CrossRef - Das, K.; Lorena, P. D.; Ng, L. K.; Lim, D.; Shen, L.; Siow, W. Y.; Teh, M.; Reichardt, J. K.; Salto-Tellez, M. Differential expression of steroid 5α-reductase isozymes and association with disease severity and angiogenic genes predict their biological role in prostate cancer. Endocr Relat Cancer 2010, 17 (3), 757-770. DOI: 10.1677/erc-10-0022.
CrossRef - Golbano, J. M.; Lóppez-Aparicio, P.; Recio, M. N.; Pérez-Albarsanz, M. A. Finasteride induces apoptosis via Bcl-2, Bcl-xL, Bax, and caspase-3 proteins in LNCaP human prostate cancer cell line. Int J Oncol 2008, 32 (4), 919-924. DOI: 10.3892/ijo.32.4.919
CrossRef
Accepted on: 18 May 2026







ISSN Online: 2231-5039

![]()




{kind=link}