Development and Validation of a Dual HPLC Method for Simultaneous Determination of Lacosamide and Methyl Paraben in Oral Solution
 , M.Sumithraand K. Archana*
, M.Sumithraand K. Archana*Department of pharmaceutical chemistry and analysis, School of Pharmaceutical science, VELS institute of science, Technology and Advanced Studies, Pallavaram, Tamil Nadu, India.
Corresponding Author E-mail:archana.drf@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/420305
Download this article as:
![]()
A rapid and stability-indicating dual high-performance liquid chromatography (HPLC) method was developed and validated for the simultaneous determination of Lacosamide and Methyl Paraben in oral solution. Chromatographic separation was achieved using a phosphate buffer of pH 3.0 (mobile phase-A) and a mixture of methanol and acetonitrile (5:95, v/v; mobile phase-B) on an Agilent Eclipse XDB-C8 column (3.5 μm, 150 × 4.6 mm). The flow rate was maintained at 1.5 mL/min, and detection was performed at 215 nm. The method exhibited excellent linearity over the concentration range of 10–150% of the working levels, with correlation coefficients (r²) of 0.9992 for Lacosamide and 0.9994 for Methyl Paraben. The percentage relative standard deviation for precision studies was less than 2.0%, confirming method reproducibility. Recovery studies indicated accuracy within the range of 98–102%. Forced degradation under acid, base, oxidative, thermal, and photolytic conditions demonstrated that the method is specific and stability-indicating, with no interference from degradation peaks. The robustness of the method was verified by small deliberate variations in flow rate and mobile phase composition. The proposed dual HPLC method is precise, accurate, and reliable for the routine quantitative estimation of Lacosamide and its preservative content in oral pharmaceutical formulations, offering an improved approach compared with the existing USP monograph.
KEYWORDS:Dual HPLC; Forced Degradation; Lacosamide; Methyl Paraben; Method Validation; Pharmaceutical Analysis.
Introduction
Lacosamide is an anticonvulsant drug recommended for the epilepsy treatment. Its IUPAC is (2R)-2-(acetylamino)-Nbenzyl-3-methoxypropanamide (C13H18N2O3) as per the Figure 1. The chemical structure of Lacosamide consists of a central amino acid core, with a functional amide group (-CONH) and a benzyl group (C6H5CH2) attached to the nitrogen atom. Additionally, Lacosamide features an ethyl chain with a hydroxyl group (-OH) at one end, which enhances its overall stability and bioavailability. Lacosamide is an antiepileptic drug that enhances the slow inactivation of voltage-gated sodium channels, thereby stabilizing hyperexcitable neuronal membranes and reducing seizure activity. It exhibits high oral bioavailability (>90%), predictable pharmacokinetics, and minimal cytochrome P450 metabolism, making it suitable for chronic therapy. Lacosamide is classified as a Biopharmaceutics Classification System (BCS) Class I drug, indicating high solubility and permeability, and is formulated as an oral solution containing methyl paraben as a preservative.1-7
Despite the availability of several analytical methods for Lacosamide determination, the United States Pharmacopeia (USP) pending monograph for Lacosamide oral solution lacks the capability to elute and quantify methyl paraben, which is essential for ensuring preservative effectiveness and product safety. Furthermore, some previously reported HPLC methods rely on trifluoroacetic acid (TFA)-based mobile phases that may compromise compound stability and interfere with UV detection due to their strong absorbance. Hence, there is a clear analytical gap in developing a single, stability-indicating method that can simultaneously estimate both Lacosamide and methyl paraben with high accuracy, sensitivity, and reproducibility.8-11
The present study aims to develop and validate a dual HPLC method for the simultaneous estimation of Lacosamide and Methyl Paraben in oral solution, using a phosphate buffer-based mobile phase that ensures better peak resolution and analyte integrity compared with TFA-based systems. The method was validated according to the International Council for Harmonisation (ICH) Q2(R1) guidelines for parameters including specificity, linearity, accuracy, precision, robustness, and solution stability. Forced degradation studies were also conducted under acid, base, oxidative, thermal, and photolytic conditions to confirm the stability-indicating nature of the method.
This validated approach provides a reliable and environment-friendly analytical alternative for routine quality control of Lacosamide oral formulations containing preservatives. It not only addresses the limitations of the existing USP method but also contributes to the advancement of dual-analyte HPLC methodologies in pharmaceutical analysis.
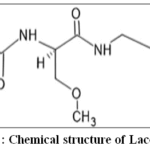 |
Figure 1: Chemical structure of Lacosamide Click here to View Figure |
Materials and Methods
Materials
Anhydrous potassium dihydrogen phosphate and orthophosphoric acid of analytical reagent (AR) grade were procured and used for buffer preparation. Methanol and acetonitrile of HPLC grade, and Milli-Q water, were used as solvents throughout the study. Lacosamide reference standard was kindly provided by MSN Pharma Chem Private Limited (Hyderabad, India). Methyl Paraben of analytical grade was purchased from a certified supplier. All chemicals and reagents used were of high purity and met analytical grade specifications.
Equipment
Chromatographic analysis was carried out using a high-performance liquid chromatography (HPLC) system equipped with a UV detector and operated with Empower software (Waters, USA). An Agilent Eclipse XDB-C8 column (150 × 4.6 mm, 3.5 μm particle size) was employed for separation. Sonication was performed using a Branson ultrasonic cleaner, and pH adjustments were made using a Calibrated pH meter (Eutech Instruments). All weighing operations were conducted on an analytical balance with 0.1 mg precision (Sartorius, Germany).
Optimization of the Mobile Phase
An accurately weighed quantity of 2.68 g of anhydrous potassium dihydrogen phosphate was dissolved in 1 L of Milli-Q water with the aid of sonication to prepare the buffer solution. The pH of the solution was adjusted to 3.0 ± 0.05 using dilute orthophosphoricacid, followed by filtration through a 0.45 μm nylon membrane filter before use.
The mobile phase consisted of two components:
Mobile Phase A: 100% phosphate buffer (pH 3.0)
Mobile Phase B: A mixture of methanol and acetonitrile in the ratio of 5:95 (v/v)
Both components were mixed well and degassed before use. The composition of the mobile phase during the gradient elution was optimized to achieve good resolution, sharp peak symmetry, and appropriate retention times for both Lacosamide and Methyl Paraben.
Optimization of chromatographic condition
Chromatographic separation was achieved on an Agilent Eclipse XDB-C8 column (150 × 4.6 mm, 3.5 μm) maintained at a column temperature of 30 °C. The mobile phase was delivered in gradient elution mode at a flow rate of 1.5 mL min⁻¹. The injection volume was 20 µL, and detection was performed using a UV detector set at 215 nm.
The optimized gradient program for mobile phases A and B is summarized in Table 1. Under these conditions, the average retention times were approximately 6 min for Lacosamide and 10 min for Methyl Paraben, providing well-resolved and symmetric peaks suitable for quantitative analysis.
Table 1: Composition of eluent varies throughout time
|
Time in minutes |
MP- A (%v/v) | MP- B (%v/v) |
| 0.0 | 85 |
15 |
|
9.5 |
75 | 25 |
| 14.00 | 65 |
35 |
|
14.01 |
85 | 15 |
| 18.00 | 85 |
15 |
Sample Preparation
Diluent preparation
Mixture of Water & Acetonitrile in a 75:25 %v/v ratio.
Standard preparation
Preparation of lacosamide standard stock solution: (Concentration about 1000 ppm).
Weigh out 25.0 mg of Lacosamide standard precisely, then transfer it to a 25 mL volumetric flask with a clear and dehydrated. After that, add 1-20% diluent, then sonicate and mack up with diluent to volume and mix evenly.
Preparation of Methyl Paraben Standard Stock Solution: (Concentration about 250 ppm)
Measure out 25.0 mg of the Methyl Paraben standard precisely, then transfer it to a 100 mL volumetric flask with a clear and dehydrated. After that, add 25-30% diluent, then sonicate and mack up with diluent to volume and mix evenly.
Preparation of standard solution
(Lacosamide concentration about 200 ppm, Methyl Paraben concentration about 50 ppm)
Prepare a 25 mL volumetric flask with 5 mL of the above Lacosamide standard stock solution and 5 mL of the above Methyl Paraben standard stock solution. Dilute with diluent to volume and well mix. Figure 3 displays the produced chromatogram, while Table 2 reports the outcome.
Sample preparation
For strength: 10 mg/mL with Methyl Paraben: 2.6 mg/mL. Figure 4 displays the produced chromatogram, while Table 3 reports the outcome.
Preparation of Lacosamide Assay: (Lacosamide concentration about 200 ppm)
Weigh out around 0.5 g of the material and pour it into a 25 ml volumetric flask that has been dehydrated. After that, it has to be well dissolved and mack up with diluent to volume. A nylon membrane filter with a pore size of 0.45 μm must be used to filter the solution.
Preparation of Methyl Paraben Assay: (Methyl Paraben concentration about 52 ppm)
After weighing out around 1 g of the material, transfer it to a well dehydrated 25 ml volumetric flask. It must then be well dissolved and mack up to volume using diluent. A nylon membrane filter with a pore size of 0.45 μm must be used to filter the solution.
System Suitability Parameters
The following system suitability parameters have been derived from the method development data for this procedure.
The USP Tailing Factor for Lacosamide and Methyl Paraben peak from the first injection of the standard solution should not exceed 1.5.
The percentage relative standard deviation of Lacosamide peak areas and Methyl Paraben peak areas from five replicate injection of standard solution should not exceed 1.0 & 2.0 respectively.
 |
Figure 2: Chromatogram of the blank sample for the assay Click here to View Figure |
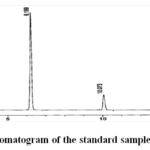 |
Figure 3: Chromatogram of the standard sample for the assay Click here to View Figure |
Table 2: Results of standard chromatogram for assay.
|
Peak |
Name | RT[Min] | Area | Height |
| 1 | Lacosamide | 6.199 | 4482.294 |
715.389 |
|
2 |
Methyl paraben | 10.073 | 2560.203 | 330.382 |
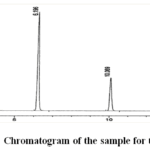 |
Figure 4: Chromatogram of the sample for the assay Click here to View Figure |
Table 3: Results of sample chromatogram for assay
|
Peak |
Name | RT[Min] | Area | Height |
| 1 | Lacosamide | 6.196 | 4397.483 |
701.246 |
|
2 |
Methyl paraben | 10.069 | 2544.697 |
330.802 |
Experimental Design
Method Development
Specificity
Solutions of Lacosamide test sample (Control Sample) and Lacosamide sample spiked with all known related substances at the specified or appropriate detection level (Spiked Sample) were prepared and injected into HPLC system according to the established methodology. Peak purity was assessed using the system software.13
Forced Degradation
The condition listed below were applied to the Lacosamide sample and its matched placebo and solutions were made and injected into the HPLC in accordance with protocol. Degradation like Acid, Base, Peroxide, Thermal and Sunlight.
Linearity
Analytical methods demonstrating linearity for both Lacosamide and Methyl Paraben ranging from 10% to 150% of the working concentration, are capable of yielding test results that are directly proportional to reflect the analyte concentration in samples within a defined range. Precision was assessed at both lower and high concentration levels, and linearity must be validated across a minimum of six distinct levels.14
Precision
The precision of an analytical procedure refers to the degree of agreement among a series of measurements obtained from multiple samplings of the same homogeneous sample, conducted under specified conditions.
Accuracy
The accuracy of an analytical method must be established across its entire range by analysing at least three different concentration levels. Accuracy is assessed by spiking the appropriate standard or API solution into placebo at 50%, 100% & 150% of the working concentration. The accuracy levels are determined accordance with the established linearity levels.
Limit of Detection (LOD), Limit of Quantitation (LOQ) Value and Range
LOD, LOQ value and Range of analytical method can be obtained from the linearity and accuracy data. Report the range in percentage in with respect to test concentration.
Solution Stability
Place the standard and sample preparation in the auto-sampler and inject the solutions at specified intervals, starting from the initial hour and continuing up to 24th hour mark. Perform a single injection of the standard and duplicate injections of samples. Calculate the percentage assay of lacosamide and preservative content at each time interval .15-18
Robustness
The robustness of an analytical procedure refers to its ability to remain unaffected by small, deliberate variations in method parameters, providing an indication of its reliability under normal operating conditions. In this experiment, method parameters are intentionally altered one at a time, and the resulting data is assessed to detect any minor changes that may occur inadvertently during routine analysis.19,20
Results
Specificity Data
Specificity was assessed by measuring interference with Blank/Diluent & placebo. The HPLC system was injected with diluent, placebo, standard preparation and sample preparation. As a result, no interference was observed at RT as shown in Table 4.
Retention Time of Lacosamide in Standard and Test were comparable
Retention Time of Lacosamide in Standard: 20 min
Retention Time of Lacosamide in Test: 18 min
Retention Time of Methyl paraben in Standard: 17 min
Retention Time of Methyl paraben in Test: 15 min
Acceptance Criteria: Results in terms of Retention Time should be equivalent to Standard.
Table 4: Difference in assay were compared
|
Sample ID |
%Assay | % Assay difference |
| Control sample | 99.5 |
0.6 |
|
Spiked sample |
98.9 |
Forced Degradation Data
The conditions listed below were applied to the lacosamide sample and its matched placebo and solutions were made and injected into the HPLC in accordance with protocol. The peak purity data for the Lacosamide peak in each degradation sample demonstrate that the peak is homogeneous, with no co-eluting peaks observed. This indicates that the method is both stability-indicating and specific. The reported data is on Table 5.
Table 5: Degradation Data for Lacosamide
|
Degradation Mechanism |
Degradation condition | Assay [%] | [%]
Degradation |
Lacosamide Peak Purity
[Main peak] |
||
| Purity
Factor |
Purity
Threshold |
Peak Purity |
||||
|
NA |
Undegraded
sample |
99.20 | NA | 999.941 | 990.00 | Pass |
| Acid | 1 N HCL /5 mL
/ 60° C/6 Hrs |
96.30 | 2.9 | 999.992 | 990.00 |
Pass |
|
Base |
0.5 N NaOH
/5 mL/60° C /6 Hrs |
97.10 | 2.1 | 999.943 | 990.00 | Pass |
| Peroxide | 3% H₂O₂ /5
mL / Bench top/ 24 Hrs |
98.90 | 0.300 | 999.990 | 990.00 |
Pass |
|
Thermal |
60° C/ 24 Hrs | 98.99 | 0.210 | 999.991 | 990.0 | Pass |
| Sunlight | Day light & UV light/3
days |
99.10 | 0.100 | 999.990 | 990.00 |
Pass |
Acceptance Criteria: There shouldn’t be any co-eluting peaks and the lacosamide peak should be uniform. Peak Purity should meet the requirements for acceptance.
Linearity Data
Using the Lacosamide working standard, a number of solutions were made with concentrations ranging from 10% to 150% was examined. Lacosamide has a 6-point calibration curve with concentrations ranging from 19.965 to 299.672µg/mL. The response rate was linear over the indicated range. The calibration equation is: y = 20.759 x + 184.27, R2 = 0.9992. Figure 5 shows the plotted graph and the data presented in Table 6. Whereas, the 6-point calibration curve for methyl paraben linearity data shows values ranging from 5.02 to 75.279 µg/mL, over the specified range the response rate was linear. R2 = 0.9994 and y = 49.682 x + 33.283 are the calibration equations. Additionally, the limit of detection and limit of quantification were found using the recommended approach in accordance with the ICH guidelines. The plotted graph and the data from Table 7 are displayed in Figure 6.
Table 6: Linearity data of lacosamide for assay
|
% Concentration [Approx.] |
Concentration
[µg/mL] |
Average area | Statistical analysis | |
| 10 | 19.965 | 574.687 | Slope |
20.7 |
|
50 |
99.964 | 2249.537 | Intercept | 184.2 |
| 75 | 149.886 | 3289.007 | % Y- Intercept |
4.2 |
|
100 |
199.768 | 4395.230 | Correlation
coefficient[r] |
0.9992 |
| 125 | 249.850 | 5438.120 | LOD |
10.06 |
|
150 |
299.672 | 6315.066 | LOQ |
31.04 |
Linearity Plot [Concentration Vs Response]
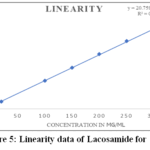 |
Figure 5: Linearity data of Lacosamide for assay Click here to View Figure |
Acceptance Criteria: A correlation coefficient of 0.9990 or above must be achieved.
Table 7: Linearity data of Methyl Paraben for assay
|
% Concentration [Approx.] |
Concentration
[µg/mL] |
Average area | Statistical analysis | |
| 10 | 5.02 | 284.212 | Slope |
49.7 |
|
50 |
25.089 | 1288.989 | Intercept | 33.2 |
| 75 | 37.639 | 1863.24 | % Y-
Intercept |
1.2 |
|
100 |
50.2 | 2542.361 | Correlation
Coefficient [r] |
0.9994 |
| 125 | 62.74 | 3194.814 | LOD |
1.95 |
|
150 |
75.279 | 3742.992 | LOQ |
5.93 |
Linearity Plot [Concentration Vs Response]
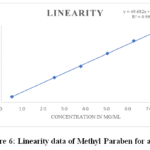 |
Figure 6: Linearity data of Methyl Paraben for assay Click here to View Figure |
Acceptance Criteria: There should be not less than 0.9990 for the correlation coefficient.
Method Precision Data
The HPLC system were given injections of six sample batches. Results are displayed below in Tables 8 & 9 for the precision data on Lacosamide and Methyl paraben, the percentage RSD of six test result was less than 2.0%. Hence, the method was precise and suitable.
Table 8: Precision Data of Lacosamide for assay
|
Sample ID |
% Assay |
|
1 |
99.1 |
| 2 |
100.6 |
|
3 |
101.3 |
| 4 |
101.7 |
|
5 |
98.1 |
| 6 |
99.3 |
|
Mean |
100.01 |
| SD |
1.4034 |
|
% RSD |
1.4 |
Acceptance Criteria: RSD as a percentage shouldn’t exceed 2.0%.
Table 9: Precision Data of Methyl Paraben for assay
|
Sample ID |
% Assay |
| 1 |
93.2 |
|
2 |
93.5 |
| 3 |
93.7 |
|
4 |
95.6 |
| 5 |
94.6 |
|
6 |
93.2 |
| Mean |
93.97 |
|
SD |
0.95 |
| % RSD |
1.0 |
Acceptance Criteria: RSD as a percentage shouldn’t exceed 2.0%.
Accuracy Data
Using a drug substance and a placebo in accordance with the test procedure, solutions were made in a single injection at concentrations ranging from 50% to 150% of the test concentration. Tables 10&11 below show that the recovery results indicating that the test method has an acceptable level of accuracy for the assay of Lacosamide and preservative content of Methyl Paraben in Lacosamide oral solution from 10% to 150% of test concentration. The requirements for system suitability were achieved. For the assay, the recovery percentage ranged from 98.0 to 102.0% and for the preservative content, it exceeded 90%.
Table 10: Accuracy data of Lacosamide for assay
|
Concentration / Sample Id |
% Recovery |
| 50% Level Sample |
101.0 |
|
100% Level Sample |
99.8 |
| 150% Level Sample |
101.3 |
Acceptance Criteria: The range for the mean recovery is 98.0% to 102.0%.
Table 11: Accuracy data of Methyl Paraben for Assay
|
Concentration / Sample Id |
% Recovery |
|
50% Level Sample |
98.1 |
| 100% Level Sample |
101.2 |
|
150% Level Sample |
99.7 |
Acceptance Criteria: Recovery should NLT 90%
Stability Data of Solution
Both standard and sample solutions were made in accordance with the test procedure, and they were kept at room temperature [~25° C] for initial analysis as well as subsequent analyses at various intervals. At room temperature, the percentage difference between assay results for Methyl Paraben and Lacosamide was shown to be stable for up to 24 hours. Tables 12 and 13 provide the data for the Lacosamide assay and Preservative content, respectively.
Table 12: Stability Data of Solution for Lacosamide for Assay
|
Room Temperature [~25° C] |
Standard | Sample | ||
| Time in hours | Area | % Difference | Area |
% Difference |
|
Initial |
4422.700 | NA | 4412.000 | NA |
| 6 Hours | 4433.000 | -0.2 | 4412.200 |
0 |
|
9 Hours |
4442.200 | -0.4 | 4415.400 | -0.1 |
| 12 Hours | 4462.290 | -0.8 | 4427.128 |
-0.3 |
|
24 Hours |
4492.342 | -1.5 | 4452.522 | -0.9 |
Acceptance criteria: The percentage difference between the regions measured at the beginning and various time intervals shouldn’t be greater than 2%.
Table 13: Stability Data of Solution for Methyl Paraben for Assay
|
Room Temperature [~25° C] |
Standard | Sample | ||
| Time in hours | Area | % Difference | Area |
% Difference |
|
Initial |
2584.451 | NA | 2572.779 | NA |
| 6 Hours | 2598.809 | -0.5 | 2581.93 |
-0.3 |
|
9 Hours |
2600.221 | -0.6 | 2583.5 | -0.4 |
| 12 Hours | 2602.756 | -0.7 | 2584.922 |
-0.4 |
|
24 Hours |
2628.331 | -1.7 | 2603.62 |
-1.2 |
Acceptance criteria: The percentage difference between the regions measured at the beginning and various time intervals shouldn’t be greater than 2%.
Robustness Data
The following intentional adjustments were made to operational and instrumental parameters:
Adjust the flow rate by 5 ml/ min ± 0.1 ml/min.
Adjust the mobile phase ratio by ± 5.
Table 14 below displays the result of the robustness data for assay. There is no discernible variation in the Lacosamide assay readings across all variable conditions that exceeds outside of the acceptable range. This experiment concludes that the method was robust and was suitable for routine analysis.
Table 14: Robustness Data for Assay
|
Parameter |
Used Condition | Altering the condition | Deviation in
% RSD |
Tailing Factor |
|
Rate of flow [ml/min] |
1.5 ml/ min ±
0.1 ml/min |
1.4 ml/ min | 0.37 |
1.09 |
| 1.6 ml/ min | 0.56 |
1.11 |
||
|
Mobile phase ratio [Buffer]: [ACN: MEOH] |
75:25 ± 5 | 80:20 | 1.02 | 1.14 |
| 70:30 | 1.07 |
1.11 |
Acceptance criteria: Absolute difference between assay values of Lacosamide at each variable condition must be not more than 2.0%.
Evaluation of System Suitability Data
Throughout the analytical method validation study, the system appropriateness of the analytical technique was examined in accordance with the specifications provided in the analytical method validation procedure, and it was determined to be acceptable. Under every validation parameter, the standard deviation and relative standard deviation meet the acceptance criteria. The result are given below in Tables 15&16.
Table 15: Evaluation Data of System Suitability of Lacosamide for Assay
|
Injection ID |
Method
Development |
Forced
Degradation |
Method Precision,
Accuracy |
Linearity, Specificity | Solution Stability | |
| Set-1 | Set-2 | |||||
| Area | Area | Area | Area | Area |
Area |
|
|
1. |
4138.238 | 4276.122 | 4255.157 | 4474.333 | 4473.596 | 4543.4 |
| 2. | 4139.101 | 4276.129 | 4261.111 | 4483.245 | 4474.667 |
4543.7 |
|
3. |
4138.121 | 4280.488 | 4254.311 | 4474.265 | 4474.488 | 4545.1 |
| 4. | 4140.201 | 4282.022 | 4255.211 | 4477.669 | 4477.442 |
4543.1 |
|
5. |
4138.789 | 4284.822 | 4258.325 | 4476.658 | 4482.255 | 4553.5 |
| Mean | 4138.89 | 4279.92 | 4256.82 | 4477.23 | 4476.48 |
4545.76 |
|
SD |
0.83 | 3.79 | 2.84 | 3.67 | 3.53 | 4.39 |
| %RSD | 0.02 | 0.08 | 0.05 | 0.1 | 0.1 |
0.1 |
|
Tailing factor |
1.07 | 1.16 | 1.10 | 1.14 | 1.16 | 1.21 |
| Theoretical
plates |
24401 | 25050 | 24433 | 22726 | 22726 |
22726 |
Acceptance Criteria
The Lacosamide peak’s tailing factor from the standard solution ought not to surpass 1.5.
The Lacosamide peak regions’ percentage RSD from five duplicate injections of the standard solution ought not to surpass 1.0.
Table 16: Evaluation Data of System Suitability of Methyl paraben for Assay
|
Injection ID |
Method
Development |
Method Precision,
Accuracy |
Linearity, Specificity | Solution Stability |
| Area | Area | Area |
Area |
|
|
1. |
2432.886 | 2552.141 | 2552.141 | 2552.141 |
| 2. | 2433.312 | 2554.121 | 2554.121 |
2554.121 |
|
3. |
2434.386 | 2554.866 | 2554.866 | 2554.866 |
| 4. | 2438.105 | 2559.311 | 2559.311 |
2559.311 |
|
5. |
2441.001 | 2560.211 | 2560.211 | 2560.211 |
| Mean | 2435.938 | 2556.13 | 2556.13 |
2556.13 |
|
SD |
2.210 | 1.845 | 1.845 | 1.845 |
| %RSD | 0.1 | 0.13 | 0.13 |
0.13 |
|
Tailing Factor |
1.00 | 1.14 | 1.14 | 1.14 |
| Theoretical
Plates |
43325 | 40896 | 40896 |
40896 |
Acceptance Criteria
The Lacosamide peak’s tailing factor from the standard solution ought not to surpass 1.5.
The Lacosamide peak regions’ percentage RSD from five duplicate injections of the standard solution ought not to surpass 2.0.
Discussion
Table 17: Comparison of chromatographic parameters of USP pending monograph Vs Current Method
| Criteria | USP Pending Monograph 43(6) | Current Method |
| Chromatographic capability | No proven ability to analyse both lacosamide and preservative content in a single chromatogram |
The current method is more advanced as it enables the analysis of both components in one chromatogram |
|
Injection volume |
Injection volume is 4 µL | Injection volume is 20 µL, providing better peak sensitivity |
| Mobile phase composition | Trifluoroacetic acid (TFA) is used as solution A and ACN is used as solution B |
Phosphate buffer used as solution A which is more suitable for analysing sensitive compounds while ACN: MeOH mixture offers better resolution and separation |
|
Peak resolution |
Limited separation and resolution due the use of TFA and ACN. | Improved resolution and separation due to the use of phosphate buffer and ACN: MeOH |
| Impact on sample integrity | Strong acidity from TFA can potentially degrade sensitive compounds |
The phosphate buffer is gentler on sensitive compounds, preserving their integrity. |
|
UV Absorbance interference |
TFA has strong UV absorbance, which may interfere with UV detection. |
The phosphate buffer has minimal UV absorbance, reducing interference. |
The current method represents a significant improvement over the existing method by addressing key limitations such as peak sensitivity, resolution and sample integrity. By enabling the analysis of both lacosamide and preservative content in a single chromatogram, using a gentler mobile phase, and reducing UV interference, it offers enhanced accuracy and reliability for chromatographic analysis (Table 17). These innovations make the current method more efficient and suitable for precise, high-quality analytical result.
Conclusion
The study successfully developed and validated a high-performance liquid chromatography method for the simultaneous quantification of Lacosamide and Methyl Paraben in pharmaceutical formulations. The method demonstrated excellent specificity, linearity, and accuracy, with correlation coefficients exceeding the accepted standard of 0.9990. Precision assessment showed minimal variability, with %RSD values below 2.0 and tailing factors consistently under 1.5, confirming peak integrity. Accuracy, evaluated through recovery studies, was within the acceptable range of 98.0–102.0% (Table 18).
Robustness testing confirmed the method’s reliability, as minor changes in flow rate and mobile phase composition had negligible impact on assay performance. Forced degradation studies provided valuable insight into the stability of Lacosamide under various stress conditions.
Overall, the validated high-performance liquid chromatography method is precise, specific, and suitable for routine quality control analysis of Lacosamide and Methyl Paraben in oral formulations. This work establishes a standardized analytical approach that can serve as a reference for future studies in pharmaceutical analysis.
Table 18: Summary of Validation Parameters
|
Parameter |
Lacosamide Assay Data | Methyl Paraben Assay Data | ACCEPTANCE CRITERIA |
| Slope | 20.7 | 49.7 |
Should be consistent and indicative of linearity |
|
Correlation coefficient |
0.9992 | 0.9994 | ≥ 0.9990 |
| LOD | 10.06 | 1.95 |
Should be low enough for reliable detection |
|
LOQ |
31.04 | 5.93 | Should be appropriate for quantitative analysis |
| Precision (% RSD) | 1.4 | 1.0 |
≤ 2.0% |
|
Accuracy (% Recovery) |
100.7 | 99.6 |
98.0%-102.0% |
Acknowledgement
The authors would like to deeply thank writers VISTAS – Vels Institute of Science,Technology&Advanced Studies (VISTAS), Pallavaram, Chennai-600117, Tamil Nadu,India, for their unending support for all the facilities and ideas provided, which have had adirect effect in developing the current work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Beydoun, A.; D’Souza, J.; Hebert, D.; Doty, P. Expert Rev. Neurother. 2009, 9, 33–42.
CrossRef - Vudagandla, S.; Dokku, R.; Uma Maheswari, B. N. RJPBCS 2011, 2, 1–4.
- Park, K. D.; Morieux, P.; Salome, C. H.; Cotton, S. W.; Reamtong, O.; Eyers, C.; Gaskell, S. J.; Stables, J. P.; Liu, R.; Khon, H. J. J. Med. Chem. 2009, 52, 6897–6911.
CrossRef - Guenter, K.; Tanja, S. J. Arzneimitteltherapie2009, 27, 157–162.
- Kristophe, S.; Elise, S. G.; Duck, P. K.; Pierre, M.; Robert, S.; Erica, D. M.; James, S. P.; Harold, K. J. J. Med. Chem. 2010, 53, 1288–1305.
CrossRef - Christian, T.; Roland, R.; Thomas, H.; Christian, E. Epilepsia2010, 51, 316–317.
- Aziz, S.; Salah, F.; Louis, S. J.; Armen, A.; Jeffrey, S.; David, S.; Sabine, B. J. Pain 2009, 10, 818–828.
- Xia, H. J.; Thomas, S.; Norma, S.; Zesuzsanna, W. H.; Jun, X. X. Eur. J. Pharmacol. 2006, 553, 135–140.
- Yuet, W. Pharma Times, UK National Electronic Library for Medicines 2007.
- Greenway, C.; Ratnaraj, N.; R., S.; Sander, J.; Patsalos, P. Ther. Drug Monit. 2010, 32, 448–452.
CrossRef - ICH Harmonised Guideline. Stability Testing of New Drug Substances and Products (Q1A(R2)) 2003, incorporated in: ICH Q1A(R3), International Council for Harmonisation, Geneva, Switzerland 2023.
- ICH Harmonised Guideline. Validation of Analytical Procedures: Methodology (Q2B) 1996, updated as ICH Q2(R2), International Council for Harmonisation, Geneva, Switzerland 2022.
- Seshachalam, U.; Haribabu, B.; Chandrasekhar, K. B. J. Sep. Sci. 2007, 30, 999–1004.
CrossRef - Rao, R. N.; Vali, R. M.; Ramachandra, B.; Raju, S. S. J. Pharm. Biomed. Anal. 2011, 54, 279–285.
CrossRef - Tamaro, I.; Aprile, S.; Giovenzan, G. B.; Grosa, G. J. Pharm. Biomed. Anal. 2010, 51, 1024–1031.
CrossRef - ICH Q2(R1), Validation of Analytical Procedures: Text and Methodology, International Conference on Harmonisation, Geneva, Switzerland 2005, incorporated into Q2(R2) 2022.
- Molleti, S.; Rao, V.; Jayaveera, K. N. Der Pharma Chem. 2013, 5, 81–89.
- Chakravarthy, V. K.; Gowri Shankar, D. Rasayan J. Chem. 2011, 4, 744–752.
- Ramisetti, N. R.; Kuntamukkala, R.; Lakshetti, S.; Sripadi, P. J. Pharm. Biomed. Anal. 2014, 95, 256–264.
CrossRef - Kim, S. J.; Koo, T. S.; Ha, D. J.; Baek, M.; Lee, S. K.; Shin, D. S.; Moon, H. Biomed. Chromatogr. 2012, 26, 371–376.
CrossRef
Accepted on: 17 Oct 2025
Second Review by: Dr. Ramesh Bhargawa
Final Approval by: Dr. Naeem Uddin Siddiqui







ISSN Online: 2231-5039

![]()




{kind=link}