Design, Molecular Docking, and In Silico ADME Evaluation of Novel 2-Substituted Benzimidazole Derivatives
 , Kratika Daniel, Sudha Vengurlekarand Sachin K Jain
, Kratika Daniel, Sudha Vengurlekarand Sachin K JainFaculty of Pharmacy, Oriental University, Indore, Madhya Pradesh, India
Corresponding Author E-mail: deepakkumawat.pch@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/420333
Download this article as:
![]()
This study investigates the antimicrobial potential of heterocyclic substituted 2-benzimidazole derivatives through molecular docking and in silico ADME analysis against glucosamine-6-phosphate synthase, an important enzyme involved in microbial cell wall synthesis. Because inhibition of this enzyme can suppress microbial growth, it has emerged as a promising target for the discovery of new antibacterial and antifungal agents. The designed benzimidazole derivatives were optimized using the LigPrep tool in Schrödinger Maestro 12.8, while the crystal structure of glucosamine-6-phosphate synthase (PDB ID: 1MOQ) was prepared for docking studies after structural refinement and energy minimization. Docking simulations were performed using the Glide XP protocol to evaluate the interaction pattern and binding affinity of the compounds within the enzyme active site. Several derivatives demonstrated strong binding interactions, including hydrogen bonding, hydrophobic interactions, and van der Waals contacts with important amino acid residues. Compounds 3g, 3f, and 3d exhibited comparatively better docking scores, suggesting significant affinity toward the target enzyme. Furthermore, pharmacokinetic and drug-likeness properties predicted using the SwissADME tool indicated favorable oral bioavailability and compliance with Lipinski’s rule of five. These findings highlight the potential of substituted benzimidazole derivatives as promising candidates for future antimicrobial drug development
KEYWORDS:Antimicrobial activity; Drug-likeness; Glucosamine-6- phosphate synthase; Glide XP; Novel benzimidazoles; Structure based docking
Introduction
The benzimidazole heterocycle has long been recognized as a “privileged scaffold” in medicinal chemistry—a structural class capable of binding to multiple, unrelated biological targets with high affinity. This privileged status stems from its remarkable structural isomorphism with naturally occurring purine nucleotides, allowing benzimidazole-containing molecules to effectively mimic the electronic and spatial properties of adenine and guanine. Consequently, these compounds can competitively inhibit a wide range of enzymatic systems, including DNA topoisomerases, RNA polymerases, and various kinases, while also interfering with tubulin polymerization and membrane integrity. Historically, the discovery of 5,6-dimethylbenzimidazole as a component of vitamin B₁₂ first alerted chemists to the biological relevance of this nucleus. Since then, a vast library of synthetic benzimidazoles has been developed, yielding numerous clinically successful drugs across diverse therapeutic categories. For gastrointestinal disorders, proton pump inhibitors such as omeprazole and lansoprazole—which contain a substituted benzimidazole core—revolutionized the treatment of peptic ulcers. In antiparasitic therapy, albendazole and mebendazole exploit the scaffold’s ability to bind parasite β-tubulin, thereby disrupting microtubule assembly. The antiviral era brought enviradine and maribavir, while antihypertensive candesartan and anticoagulant dabigatran further underscore the scaffold’s versatility. From a synthetic perspective, the benzimidazole ring system is highly tunable: the two nitrogen atoms at positions 1 and 3 allow for alkylation/arylation, while the benzene portion (positions 4–7) and the carbon at position 2 are readily modified via electrophilic substitution, condensation with aldehydes, or metal-catalyzed cross-coupling reactions. This synthetic accessibility facilitates rapid structure-activity relationship (SAR) exploration. In the context of antimicrobial drug discovery, benzimidazoles have regained intense interest due to escalating multidrug resistance among bacteria and fungi. Their dual mechanisms—often involving both DNA synthesis inhibition and membrane disruption—make them less prone to single-point resistance mutations. Moreover, the scaffold can accommodate electron-withdrawing groups (e.g., –Cl, –NO₂, –CF₃) and lipophilic side chains that enhance penetration through Gram-positive and Gram-negative cell envelopes. Therefore, the benzimidazole core continues to serve as an ideal starting point for designing novel antimicrobial candidates, offering a well-balanced combination of established pharmacological precedent, synthetic flexibility, and multiple points for structural diversification.
 |
Scheme 1: Benzimidazole Click here to View Scheme |
Materials and Methods
Protein Preparation
The three-dimensional crystal structure of glucosamine-6-phosphate synthase (PDB ID: 1MOQ) was retrieved from the Protein Data Bank for molecular docking studies involving synthesized 2-substituted benzimidazole derivatives. Protein preparation was carried out using the Protein Preparation Wizard available in Schrödinger Maestro 12.8. During this process, missing hydrogen atoms were incorporated, bond orders were corrected, formal charges were adjusted, and incomplete residues were refined to obtain a reliable protein structure. Unnecessary water molecules located farther than 5 Å from the active binding region were deleted, whereas functionally important water molecules were preserved.
Further optimization included correction of ionization states and refinement of the hydrogen-bonding network. The prepared protein structure was then subjected to restrained energy minimization using the OPLS force field with an RMSD cutoff value of 0.3 Å. The finalized and energetically stable protein model was subsequently employed for docking studies to investigate the binding behavior and interaction profile of the designed benzimidazole derivatives with glucosamine-6-phosphate synthase.
Ligand Preparation
The chemical structures of the synthesized 2-substituted benzimidazole derivatives were created using ChemDraw Ultra and stored in SDF format. These ligand files were imported into the LigPrep tool of Schrödinger Maestro 12.8 for further processing and optimization prior to molecular docking studies with glucosamine-6-phosphate synthase.
During ligand preparation, the 2D molecular structures were converted into energetically favorable 3D conformations. Various stereoisomers, tautomeric forms, and ionization states were generated to obtain chemically and biologically relevant ligand structures. Essential hydrogen atoms were incorporated, bond orders were adjusted, and incorrect chiral configurations were corrected to improve structural accuracy. Appropriate ionization states at physiological pH (6.8–7.2) were predicted using the Epik module.
The prepared ligands were subsequently subjected to geometry refinement and energy minimization with the OPLS force field to achieve stable low-energy conformations. The optimized benzimidazole derivatives were finally selected for docking studies to investigate their binding interactions and possible inhibitory effects against glucosamine-6-phosphate synthase.
Receptor Grid Generation
The binding site of glucosamine-6-phosphate synthase was determined with the help of the co-crystallized ligand GLP available in the protein crystal structure. Using the Glide application in Schrödinger Maestro 12.8, a receptor grid was created around the identified active site to facilitate molecular docking analysis. The dimensions of the grid box were maintained at 14 Å × 14 Å × 14 Å to ensure complete coverage of the binding cavity and to provide sufficient space for the interaction of synthesized 2-substituted benzimidazole derivatives with key amino acid residues. The generated receptor grid was then employed for subsequent docking studies to analyze ligand–protein binding behavior and inhibitory potential.
Validation of the Docking Procedure
The docking protocol was validated to ensure the correctness and consistency of the molecular docking study. This was achieved by comparing the predicted orientation of the co-crystallized ligand with its experimentally resolved crystal structure. For validation purposes, the native ligand present in the active site of glucosamine-6-phosphate synthase was first removed and then reintroduced into the same binding pocket through the Glide docking method.
The generated docked pose was carefully examined by considering docking score, interaction pattern, and orientation inside the active site region. Hydrogen-bond interactions and ligand fitting within the receptor cavity were also analyzed. The accuracy of the docking method was further determined by calculating the Root Mean Square Deviation (RMSD) between the redocked ligand and its original crystallographic conformation. A low RMSD value indicated close similarity between the predicted and experimental poses, thereby confirming the validity and reliability of the adopted docking protocol for evaluating the interaction of substituted benzimidazole derivatives with glucosamine-6-phosphate synthase.
Glide Ligand Docking
Molecular docking analysis of the synthesized 2-substituted benzimidazole derivatives was performed using the Glide module integrated in Schrödinger Maestro 12.8. The prepared receptor grid of glucosamine-6-phosphate synthase was utilized for docking calculations. Docking experiments were carried out in Extra Precision (XP) mode to investigate the binding affinity and interaction profile of the ligands within the enzyme active site.
Flexible ligand docking methodology was employed to generate multiple possible conformations for each compound. The generated poses were screened using Glide scoring functions to identify the most favorable ligand–protein interactions. During docking analysis, parameters such as hydrogen bonding, hydrophobic contacts, electrostatic interactions, and steric fitting between the ligands and active site residues were thoroughly evaluated.
The selected docked complexes were further refined through energy minimization using the OPLS force field, and the final conformations were ranked according to their XP-Glide scores. Several benzimidazole derivatives demonstrated strong binding affinity and stable interactions with crucial amino acid residues present in the active site of glucosamine-6-phosphate synthase. The docking outcomes indicated that these compounds may act as potential enzyme inhibitors and could be explored further as promising antimicrobial candidates.
Result and Discussion
Molecular docking studies of the synthesized 2-substituted benzimidazole derivatives were performed using the Schrödinger Maestro 12.8 software to examine their binding efficiency against glucosamine-6-phosphate synthase. The docking investigation was carried out to assess the possible inhibitory activity of the designed molecules toward the target enzyme. The obtained Glide scores demonstrated that several compounds interacted effectively within the active site of glucosamine-6-phosphate synthase. Certain derivatives exhibited superior docking scores due to favorable hydrophobic contacts, hydrogen-bonding interactions, and electrostatic attractions with important amino acid residues of the receptor cavity. The docking findings indicated the formation of stable ligand–protein complexes, suggesting that the synthesized benzimidazole derivatives may act as potential glucosamine-6-phosphate synthase inhibitors. The most active docked poses showed significant binding interactions through hydrogen bonds and van der Waals forces, which may contribute to their antimicrobial potential. The docking results are presented in Table 1.
Table 1: Results of docking analysis for the examined compounds against Glucosamine-6-phosphate synthase.
|
Compound name. |
Docking Score |
| 3c |
-10.342 |
|
3d |
-10.441 |
| 3f |
-10.444 |
|
3g |
-10.981 |
| 3h |
-10.162 |
|
3i |
-10.187 |
| 3j |
-10.045 |
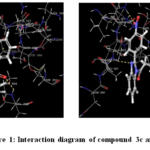 |
Figure 1: Interaction diagram of compound 3c and 3g Click here to View Figure |
The physicochemical and pharmacokinetic characteristics of the designed compounds were further evaluated through in silico ADME prediction using the SwissADME web tool. Various parameters including hydrogen bond donor and acceptor properties, lipophilicity, oral bioavailability, drug-likeness, and other ADMET-related descriptors were avourab. Most of the compounds exhibited acceptable pharmacokinetic avourabl and complied with Lipinski’s rule of five with no violations, indicating avourable drug-like properties. In addition, several derivatives demonstrated good predicted gastrointestinal absorption and oral bioavailability. Overall, the ADME analysis suggested that the synthesized 2-substituted benzimidazole derivatives possess promising pharmacological characteristics and could be considered as potential candidates for further antimicrobial studies targeting glucosamine-6-phosphate synthase. The detailed ADME data of the compounds are summarized in Table 2.
Table 2: Physicochemical and ADME characteristics of the compound were assessed through the SwissADME web tool
|
CompoundName (Code) |
Physicochemical descriptors | ADME descriptors | |||||||
| MW | NRB | NHA | NHD | TPSA | Log P o/w | Log S (ESOL) | Lipinski Rule |
Leadlikeness |
|
|
3c |
364.08 | 3 | 1 | 2 | 60.61 | 3.14 | -2.52 | 0 | 2 |
| 3d | 439.25 | 3 | 2 | 1 | 50.52 | 2.16 | -2.77 | 0 |
1 |
|
3f |
357.11 | 4 | 2 | 1 | 100.01 | 3.02 | -5.12 | 0 | 1 |
| 3g | 309.11 | 3 | 2 | 1 | 74.32 | 3.02 | -3.07 | 0 |
1 |
|
3h |
346.09 | 4 | 4 | 1 | 96.61 | 3.08 | -3.56 | 0 | 2 |
| 3i | 309.11 | 3 | 2 | 2 | 46.89 | 3.17 | -4.07 | 0 |
1 |
|
3j |
342.04 | 3 | 2 | 2 | 65.62 | 2.91 | -4.45 | 0 |
2 |
 |
Table 3: 2-substituted benzimidazole derivatives (3c- 3h). Click here to View Table |
Conclusion
The present investigation demonstrated that substituted 2-benzimidazole derivatives possess significant binding affinity toward glucosamine-6-phosphate synthase, indicating promising antimicrobial potential. Molecular docking and ADME analysis revealed favorable ligand–protein interactions and acceptable drug-like characteristics. Among the evaluated compounds, several derivatives showed superior docking performance, suggesting their suitability as lead candidates for further optimization and experimental antimicrobial studies.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports. 2017;7:42717.
CrossRef - Friesner RA, Banks JL, Murphy RB, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of Medicinal Chemistry. 2004;47(7):1739–1749.
CrossRef - Halgren TA, Murphy RB, Friesner RA, et al. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. Journal of Medicinal Chemistry. 2004;47(7):1750–1759.
CrossRef - Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Journal of Computer-Aided Molecular Design. 2013;27(3):221–234.
CrossRef - Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews. 2001;46(1–3):3–26.
CrossRef - Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence oral bioavailability of drug candidates. Journal of Medicinal Chemistry. 2002;45(12):2615–2623.
CrossRef - Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. Journal of the American Chemical Society. 1996;118(45):11225–11236.
CrossRef - Trott O, Olson AJ. AutoDockVina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry. 2010;31(2):455–461.
CrossRef - Kaur K, Jain M, Reddy RP, Jain R. Quinolines and structurally related heterocycles as antimalarials. European Journal of Medicinal Chemistry. 2010;45(8):3245–3264.
CrossRef - Kumar S, Bawa S, Gupta H. Biological activities of benzimidazole derivatives: A review. Mini Reviews in Medicinal Chemistry. 2009;9(14):1648–1654.
CrossRef
Accepted on: 13 May 2026







ISSN Online: 2231-5039

![]()




{kind=link}