Design, In Silico Studies and Molecular Docking Studies of (2E)-2-Cyano-3-Phenyl-N’-(Phthalazin-1-yl)Prop-2-enehydrazide Derivatives as Antimicrobial Agents
 , Vidya Rani Murthi1, Vanitha Madhuri Tadepalli1 and Gnaneswari Kongara2
, Vidya Rani Murthi1, Vanitha Madhuri Tadepalli1 and Gnaneswari Kongara21Institute of Pharmaceutical Technology, Sri Padmavati Mahila Visvavidyalayam, Tirupati, Andhra Pradesh, India
2Department of Applied Microbiology, Sri Padmavati Mahila Visvavidyalayam, Tirupati, Andhra Pradesh, India
Corresponding Author E-mail: grajitha@spmvv.ac.in
DOI : http://dx.doi.org/10.13005/ojc/420308
Download this article as:
![]()
The increasing number of multidrug resistant microbial infections has created a critical need for the discovery of novel antimicrobial agents. Hence, in this study, a novel series of (2E)-2-cyano-3-phenyl-N'-(phthalazin-1-yl)prop-2-enehydrazides (4a-l)were designed and in silico studies were carried out to predict molecular properties, ADME properties and toxicities of title compounds. Further, molecular docking studies were performed against S. aureus Gyrase B (PDB ID: 4URO) and E. Coli Mur B (PDB: 2Q85). In silico screening indicated all the derivatives obeyed Lipinski’s rule of five and showed favourable ADME properties. From molecular docking studies it was observed that3-methoxy-4-hydroxy derivative 4k exhibited good binding affinity with binding site of S. aureus gyrase B with XP G score -6.362 Kcal/mol compared to that of crystal ligand novobiocin (-5.964 Kcal/mol). Compound 4a showed good binding affinity with E. coli Mur B active site with docking score -6.564 Kcal/mol than that of co-crystal ligand (-6.026 Kcal/mol). From the present study, compounds 4k and 4a wereproposed to serve as promising molecules among the designed compounds for further evaluation as antibacterial agents.
KEYWORDS:Antimicrobial Activity; Hydralazine; In silico studies; Molecular docking
Introduction
Microbial infections caused by bacteria, fungi, viruses and parasites are highly contagious, resulting in serious complications impacting public health. Antibiotic resistance with current antimicrobial drugs has posed a critical challenge in the treatment of these infections, leading to a gradual increase in the frequency of microbial infections. These infections markedly cause mortality as well as morbidity, adversely affecting patients’ health and delaying their recovery. 1,2. Hence, discovery of new antibacterial agents is crucial to overcome the limitations of current therapies and also to address the multidrug-resistant infections. Cinnamamides and phthalazines are two privileged structural scaffolds that have been investigated in the search of novel antimicrobial agents.
Cinnamamideis a privileged structural scaffold in drug discovery. It exists in two geometric isomers cis and trans form. Cinnamamide derivativeshave attracted considerable interest in pharmaceutical research due to their synthetic accessibility, low toxicity profiles, and multifunctional therapeutic potential. Severalcinnamidesderivatives were reported to have diverse biologicalactivities including antimicrobial 3, antitubercular 4-6, antimalarial 7, antidiabetic 8, antiinflammatory 9,anticancer 10-12, antiviral 13-15, anticonvulsant 16, neuroprotective 17, 18, antioxidant 19 etc. Few examples of marketed drugs containing cinnamamide nucleus are Cinromide which is used as is anticonvulsant and Tranilast is an antiallergic drug 20. In addition, a significant number of patented chemical compounds were found to contain the cinnamamide nucleus.
Phthalazine, a nitrogen-containing heterocyclic compound which is also known as benzopyridazine or benzo-orthodiazine 21. Research on phtahlazine has revealed that introducing structural versatility improves its potential with regard to various pharmacological activities including anticancer 22, Anticonvulsant 23, antihypertensive 24, antimicrobial 25 activities etc. Hydralazine, a phthalazine-containing drug, has gained more attention for the synthesis of new drugs due to its pharmacological potential. Studies have shown that Hydralazine also has potent antioxidant 26, anticancer 27, antimicrobial 28, 29, and anti-aging 30 benefits.
In view of the pharmacological potential of cinnamamide pharmacophore and hydralazine, it was planned to design a novel series of cinnamoyl hydralazine derivatives as antimicrobial agents. In silico studies were carried outfor title compounds to predict their molecular properties. Further, molecular docking studies were also performed against S. aureus gyrase B (GyrB) (PDB: 4URO) and E. coli Mur B (PDB: 2Q85).
Materials and Methods
In silico studies
Molecular properties, ADME and toxicity prediction
Swiss ADME was used for prediction of various physicochemical parameters of title compounds (4a-l) that influence molecule activity such as Molecular Weight, volume, Molecular Polar Surface Area (PSA), Hydrogen bond acceptor/donar, log P and Number of rotatable bonds were calculated for the prediction of drug- likeliness of any molecule which indicates overall potential of the compound to be a drug candidate. ADME properties of title compounds were also estimated using SwissADME webtool in which, pharmacokinetic properties including GI absorption, blood brain barrier permeability and skin permeability and structural alerts were predicted31. ProTox-II was used to predict the toxicity of designed compounds, in which oral toxicity, organ toxicity, carcinogenicity, mutagenicity were predicted 32.
Molecular docking studies
Ligand preparation
Ligand (4a-l) structures were drawn using chemsketch and prepared using Epik module of Schrödinger, such as ligand’s stereochemical nature enhancement and protonation state enhancement, developing tautomers and ligand energy was minimized by using force field OPLS_3 at pH 7.0±2.033.
Protein preparation
S. aureus DNA GyrB (PDB ID: 4URO) and E. Coli MurB (PDB: 2Q85) were considered in this study to determine the binding interaction modes and binding affinities of the title compounds. 4URO represents the N-terminal domain of the S. aureus GyrB protein, a key component of bacterial DNA gyrase. 4URO is known to bind to and be inhibited by novobiocin, a coumarin antibiotic that targets the ATPase domain of gyrase. 2Q85 represents the E. coli MurB bound to naphthyl tetronic acid inhibitor (973): ((5z)-3-(4-chlorophenyl)-4-hydroxy-5-(1 naphthylmethylene)furan-2(5h)-one). This inhibitor binds to MurB’s active site, by H- bonds, to prevent its catalytic function. The target crystal structures were imported to Maestro and protein preparation was done using protein preparation wizard. Protein preparation includes hydrogen atoms addition, bond order and formal charge correction, atomic clash removal, alteration of protein tautomeric and ionization states. Hydroxyl and thiol groups of protein were reoriented to optimize the H-bonding network. Protein structures were optimized at neutral pH and energy was minimized by OPLS_3 force field for all atoms. A receptor grid was generated and undesirable water molecules were removed around inhibitor binding site of selected targets using Glide v7.1 and Protein preparation wizard respectively 34.
Molecular docking
The binding affinity between the selected targets and title compounds was determined by GLIDE-XP docking. The prepared ligands (4a-l) were docked into the grid box generated in selected target proteins using Monte Carlo-based simulated algorithm minimization method. (Gscore (Glide Score) was used to represent binding affinity34.
Results and Discussion
In silico studies
Molecular properties, ADME and toxicity prediction
All the molecular properties of the designed derivatives (4a-l) were predicted by using SwissADME web-based tool. The results suggested that all the derivatives obeyed Lipinski rule of five, which is estimated from the molecular properties such as molecular weight, H-bond acceptors and donors indicating their good oral bioavailability. Molecular properties of the title compounds was observed to be in the range of miLogP: 1.63-3.72; TPSA: 90-131; MW: 315.33-405.41; HBA:4-7; HBD:2-4; nviol:0-1; roB: 5-8. (Table 1). The results of in silico data indicates that, the title compounds may have the potential to become a lead compound. Prediction of ADME properties of title compounds by SwissADME indicated that the title compounds may exhibit high GI absorption, moderate skin permeability and bioavailability score 0.55. None of the derivatives exhibited P-glycoprotein inhibition and BBB permeation, and few of the title compounds were found to be Cytochrome P450 enzyme inhibitors (Table 2). Toxicity studies by ProTOX-II revealed that all the title compounds are inactive for cardiotoxicity, and active for hepatotoxicity, and few compounds were found to be inactive for mutagenicity (Table 3).
 |
Scheme 1: General structure of title compounds Click here to View Scheme |
Table 1: Prediction of molecular properties of title compounds
| Compound | R | Mol. wt | HBA | HBD | Nrtb | TPSA (oA) |
Log Po/w |
|
4a |
H | 315.33 | 4 | 2 | 5 | 90.7 | 2.29 |
| 4b | 4-Cl | 349.77 | 4 | 2 | 5 | 90.7 |
2.76 |
|
4c |
4-Nitro | 360.32 | 6 | 2 | 6 | 136.52 | 1.85 |
| 4d | 4-OCH3 | 345.35 | 5 | 2 | 6 | 99.93 |
2.35 |
|
4e |
3,4-(OCH3)2 | 375.38 | 6 | 2 | 7 | 109.16 | 2.42 |
| 4f | 3,4,5-(OCH3)3 | 405.41 | 7 | 2 | 8 | 118.39 |
2.00 |
|
4g |
2,4,5-(OCH3)3 | 405.41 | 7 | 2 | 8 | 118.39 | 2.44 |
| 4h | 4-OH | 331.33 | 5 | 3 | 5 | 110.93 |
1.81 |
|
4i |
3,4-(OH)2 | 347.32 | 6 | 4 | 5 | 131.16 | 1.63 |
| 4j | 3-OH | 331.10 | 5 | 3 | 5 | 110.93 |
1.97 |
|
4k |
3-OH, 4-OCH3 | 361.35 | 6 | 3 | 6 | 120.16 | 2.08 |
| 4l | 4-C6H5 | 405.41 | 4 | 2 | 6 | 90.70 |
3.72 |
*Mol. Wt: molecular weight, HBA: No. of H-bond acceptors; HBD: No. of H-bond donors; TPSA: Topological Polar Surface Area; Nrtb: No. of rotatable bonds, Log Po/w:octanol-water coefficient
Table 2: Prediction of pharmacokinetic properties of title compounds by Swiss ADME
| Compound | GI | BBB | Pgp substrate | Cytochrome P450 inhibition | ||||
| CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | ||||
| 4a | High | No | No | Yes | No | Yes | No | No |
| 4b | Yes | Yes | Yes | No | ||||
| 4c | Yes | No | Yes | No | ||||
| 4d | Yes | No | Yes | No | ||||
| 4e | Yes | No | Yes | Yes | ||||
| 4f | No | No | Yes | Yes | ||||
| 4g | No | No | Yes | Yes | ||||
| 4h | Yes | No | No | No | ||||
| 4i | Yes | No | No | No | ||||
| 4j | Yes | No | No | No | ||||
| 4k | Yes | No | Yes | No | ||||
| 4l | Yes | Yes | Yes | No | ||||
*GI absorption: Gastrointestinal absorption, BBB permeant: Blood brain barrier permeation, Pgp substrate: P-glycoprotein substrate
Table 3: Prediction of toxicity of title compounds using ProTOX-II
| Compound | Mutagenicity | Cardiotoxicity | Hepatotoxicity |
| 4a | Inactive | Inactive | Active |
| 4b | Inactive | Inactive | Active |
| 4c | Active | Inactive | Active |
| 4d | Active | Inactive | Active |
| 4e | Active | Inactive | Active |
| 4f | Active | Inactive | Active |
| 4g | Active | Inactive | Active |
| 4h | Inactive | Inactive | Active |
| 4i | Active | Inactive | Active |
| 4j | Active | Inactive | Active |
| 4k | Active | Inactive | Active |
| 4l | Inactive | Inactive | Active |
Molecular Docking
Ligand preparation
Title compounds were prepared using Ligprep with Epik module of schrodinger.
Protein preparation
S. aureus DNA GyrB (PDB: 4URO) in complex with novobiocin and E. coli MurB in complex withinhibitor 973 (PDB: 2Q85)were selected for molecular docking studies as they feature a known inhibitor binding domain which is crucial for determining the antibacterial activity of the title compounds 35,36. Inhibitor binding residues were defined around grid generated within the selected target. S. aureus GyrB and novobiocin complex binding site comprises residues such as Ser 55, Asp 57, Glu 58, Gly 85, Asn 54, Asp 81, Arg 144, Arg 84, Arg 200, Asp 89, Pro 87, Ile 102, Ile 86, Ser 128 within the 4 A0 surrounding co-crystallized inhibitor novobiocin. E. coli MurB and co-crystal ligand 973 complex binding site comprises residues such as Tyr 190, Leu 218, Ser 229, Asn 233, Pro 252, Tyr 254, Lys 262, Leu 263, Ala 264, gln 288, Leu 290, Val 291 within the 4 A0 surrounding co-crystal ligand 973. The binding site residues of selected targets were defined using PDBsum web server.
Molecular docking
Antibacterial activity of compounds 4a-l was determined by performing molecular docking studies to predict their ability in inhibiting S. aureus GyrB enzyme. S. aureus GyrB is a subunit of the type II topoisomerase enzyme responsible to introduce negative supercoils into DNA, which is important for DNA replication, transcription and other important cellular processes 37. Among all the docked complexes, 4k possesses greater binding affinity against S. aureus with XP Gscore -6.362 Kcal/mol of than that of crystal ligand novobiocin (-5.964 Kcal/mol) (Table 4). Analysis of docking interaction had revealed that hydroxyl substitution on cinnamoyl moiety of 4kformed one H-bond interaction with Asn 54 of S. aureus GyrB and two intermolecular H-bond interactions with active site residue Gly 85, one Pi-cation interaction with Arg 84 residues of S. aureus Gyr B (Fig 1). Co-crystal ligand Novobiocin exhibited two H-bond interactions with Gln 91 and Gln 92 residues of S. aureus Gyr B (Fig 2).From molecular docking studies, it was observed that compounds substituted with electron donating hydroxyl and methoxy groups on cinnamoyl moiety exhibited interactions with key amino acid residues in the binding site of S. aureus GyrB enzyme.
Title compounds were subjected to molecular docking with E. coli Mur B protein which plays a crucial role in biosynthesis of peptidoglycan, which is a vital component of the cell wall of bacteria.38 Among all the docked complexes, 4a possesses better binding affinity with XP Gscore of -6.564 Kcal/mol (Table 5) than that of crystal ligand (973) -6.026 Kcal/mol. Compound 4a exhibited H-bond interactions with Arg 159 and Glu 325 amino acid residues of E. coli Mur B (Fig 3). Compound 973 formed two hydrogen bond interactions with Gly 123 and Ser 229 residues of E. coli Mur B (Fig 4). Molecular docking studies suggested that most of the compounds in the series exhibited key interactions in the binding site of E. coli Mur B. Particularly unsubstituted derivative, para substitution with electron withdrawing chloro group and electron donating hydroxyl groups showed good binding affinity with the active site of E. coli Mur B.
Table 4: Docking results of S. aureus GyrB with title compounds (4a-l)
| Compound | R | XP G scores (Kcal/mol) | Interacting amino acids |
| 4a | H | -5.496 | – |
| 4b | 4-Cl | -5.042 | – |
| 4c | 4-Nitro | -4.526 | – |
| 4d | 4-OCH3 | -4.198 | Arg 144 |
| 4e | 3,4-(OCH3)2 | -5.319 | – |
| 4f | 3,4,5-(OCH3)3 | -4.408 | Arg 144 |
| 4g | 2,4,5-(OCH3)3 | -4.391 | – |
| 4h | 4-OH | -4.816 | – |
| 4i | 3,4-(OH)2 | -5.153 | Asp 81, Gly 85 |
| 4j | 3-OH | -4.261 | – |
| 4k | 3-OH, 4-OCH3 | -6.362 | Gly 85, Asn 54, Arg 84 |
| 4l | 4-C6H5 | -4.386 | – |
| novobiocin | -5.964 | Gln 91, Gln 92 | |
Table 5: Docking results of E. coli Mur B with title compounds (4a-l)
| Compound | R | XP G scores (Kcal/mol) | Interacting amino acids |
| 4a | H | -6.564 | Arg 159, Glu 325 |
| 4b | 4-Cl | -6.414 | Arg 159, Glu 325 |
| 4c | 4-Nitro | -6.099 | Lys 217, Arg 159, Glu 325, Gln 288 |
| 4d | 4-OCH3 | -6.046 | Arg 159, Glu 325 |
| 4e | 3,4-(OCH3)2 | -6.263 | Arg 159, Glu 325 |
| 4f | 3,4,5-(OCH3)3 | -5.822 | Gly 123, Lys 262, Tyr 190 |
| 4g | 2,4,5-(OCH3)3 | -5.681 | – |
| 4h | 4-OH | -6.500 | Arg 159, Glu 325 |
| 4i | 3,4-(OH)2 | -6.396 | Tyr 125, Arg 159, Glu 325 |
| 4j | 3-OH | -5.814 | – |
| 4k | 3-OH, 4-OCH3 | -5.690 | – |
| 4l | 4-C6H5 | -5.456 | – |
| 973 | -6.026 | Gly 123, Ser 229 | |
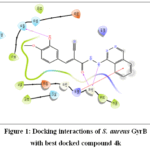 |
Figure 1: Docking interactions of S. aureus GyrB with best docked compound 4k Click here to View Figure |
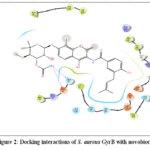 |
Figure 2: Docking interactions of S. aureus GyrB with novobiocin Click here to View Figure |
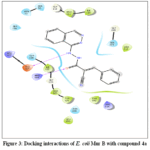 |
Figure 3: Docking interactions of E. coli Mur B with compound 4a Click here to View Figure |
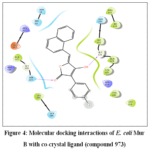 |
Figure 4: Molecular docking interactions of E. coli Mur B with co-crystal ligand (compound 973). Click here to View Figure |
Conclusion
In present study, design. in silico investigation and molecular docking studies were carried out for novel (2E)-2-cyano-3-phenyl-N‘-(phthalazin-1-yl)prop-2-enehydrazides for their antibacterial activity (4a-l). Molecular properties, ADME properties and toxicities were predicted by Swiss ADME and ProTOX-II respectively. Further, molecular docking studies were performed against S. aureus GyrB (PDB ID: 4URO) and E. coli Mur B ((PDB ID: 2Q85).All the derivatives obeyed Lipinski rule of five, indicating drug-likeness of title compounds. Pharmacokinetic properties prediction by Swiss ADME revealed that all the compounds may exhibit good GI absorption. Among all the designed compounds, 4k possesses greater binding affinity with S. aureus (-6.362 Kcal/mol) and compound 4a possesses better binding affinity with E. coli (-6.564 Kcal/mol) compared to respective standards. The results of in silico studies indicates that, the title compounds may have the potential to become a lead compound. Further studies needed for optimization of the antimicrobial properties of the title compounds.
Acknowledgement
The authors gratefully acknowledge the funding of the Pradhan Mantri Uchchatar Shiksha Abhiyan (PM-USHA), under the Multi-Disciplinary Education and Research Universities (MERU) Grant sanctioned to Sri Padmavati Mahila Visvavidyalayam, Tirupati for carrying out this research work.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Chinemerem, N. D.; Ugwu, M.C.; Oliseloke Anie C.; Al-Ouqaili, M.T.S.; Chinedu, I. J.; Victor, C. U.; Saki, M. Clin. Lab. Anal.2022, 36(9), e24655. https://doi.org/10.1002/jcla.24655.
CrossRef - Galla, R.; Puranam, L.M. J. Chem.2025, 41(1), https://dx.doi.org/10.13005/ojc/410116.
CrossRef - Narasimhan, B.; Belsare, D.; Pharande, D.; Mourya, V.; Dhake, A. J. Med. Chem.2004, 39 (10), 827-834, https://doi.org/10.1016/j.ejmech.2004.06.013.
CrossRef - Bairwa, R.; Kakwani, M.; Tawari, N. R.; Lalchandani, J.; Ray, M. K.; Rajan, M. G. R.; Degani, M. S. Med. Chem. Lett.2010, 20(5), 1623-1625, https://doi.org/10.1016/j.bmcl.2010.01.031.
CrossRef - Kakwani, M. D.; Suryavanshi, P.; Ray, M.; Rajan, M. G. R.; Majee, S.; Samad, A.; Devarajan, P.; Degani, M. S. Med. Chem. Lett. 2011,21(7), 1997-1999, https://doi.org/10.1016/j.bmcl.2011.02.022.
CrossRef - Yoya, G. K.; Bedos-Belval, F.; Constant, P.; Duran, H.; Daffé, M.; Baltas, M. Med. Chem. Lett. 2009,19(2), 341-343, https://doi.org/10.1016/j.bmcl.2008.11.082.
CrossRef - Pérez, B. C.; Teixeira, C.; Albuquerque, I. S.; Gut, J.; Rosenthal, P. J.; Gomes, J. R. B.; Prudêncio, M.; Gomes, P. Med. Chem. 2013,56(2), 556-567, https://doi.org/10.1021/jm301654b.
CrossRef - Phuwapraisirisan, P.; Puksasook, T.; Jong-aramruang, J.; Kokpol, U. Med. Chem. Lett. 2008, 18(18), 4956-4958, https://doi.org/10.1016/j.bmcl.2008.08.024.
CrossRef - Doherty, E. M.; Fotsch, C.; Bo, Y.; Chakrabarti, P. P.; Chen, N.; Gavva, N.; Han, N.; Kelly, M. G.; Kincaid, J.; Klionsky, L.; Liu, Q.; Ognyanov, V. I.; Tamir, R.Wang, X.; Zhu, J.; Norman, M. H.; Treanor, J. J. S. Med. Chem.2005, 48(1), 71-90, https://doi.org/10.1021/jm049485i.
CrossRef - Dothager, R. S.; Putt, K. S.; Allen, B. J.; Leslie, B. J.; Nesterenko, V.; Hergenrother, P. J. Am. Chem. Soc.2005, 127(24), 8686-8696, https://doi.org/10.1021/ja042913p.
CrossRef - Romagnoli, R.; Baraldi, P. G.; Salvador, M. K.; Chayah, M.; Camacho, M. E.; Prencipe, F.; Hamel, E.; Consolaro, F.; Basso, G.; Viola, G. J. Med. Chem. 2014,81, 394-407, https://doi.org/10.1016/j.ejmech.2014.05.028.
CrossRef - Zhang, M.; Lu, X.; Zhang, H.-J.; Li, N.; Xiao, Y.; Zhu, H.-L.; Ye, Y.-H. Chem. Res. 2013,22(2), 986-994, https://doi.org/10.1007/s00044-012-0093-z.
CrossRef - Wang, P.-C.; Chiu, D.-C.; Jan, J.-T.; Huang, W.-I.; Tseng, Y.-C.; Li, T.-T.; Cheng, T.-J.; Tsai, K.-C.; Fang, J.-M. J. Med. Chem. 2018,145, 224-234, https://doi.org/10.1016/j.ejmech.2017.12.072.
CrossRef - Wang, Z.; Xie, D.; Gan, X.; Zeng, S.; Zhang, A.; Yin, L.; Song, B.; Jin, L.; Hu, D. Med. Chem. Lett.2017, 27(17), 4096-4100, https://doi.org/10.1016/j.bmcl.2017.07.038.
CrossRef - Zhao, Y.; Gu, Q.; Morris-Natschke, S. L.; Chen, C.-H.; Lee, K.-H. Med. Chem. 2016,59(19), 9262-9268, https://doi.org/10.1021/acs.jmedchem.6b00461.
CrossRef - Balsamo, A.; Crotti, P.; Lapucci, A.; Macchia, B.; Macchia, F.; Cuttica, A.; Passerini, N. Med. Chem. 1977, 20(1), p 48-53, https://doi.org/10.1021/jm00211a009.
CrossRef - Peng, S.; Zhang, B.; Meng, X.; Yao, J.; Fang, J. Med. Chem.2015,58 (13), 5242-5255, https://doi.org/10.1021/acs.jmedchem.5b00410.
CrossRef - Wu, B.; Zhou, L.; Cai, H. H. Chem. Lett.2008, 19 (10), 1163-1166, https://doi.org/10.1016/j.cclet.2008.06.052.
CrossRef - Park, N.S.; Jung, Y.S.; Seong, C.M.; Lim, H.J.; Yoon, J.H.; Kong, J.Y.; Park, W.K. Google Patents: 2006,US7078407B2 – 4-hydroxycinnamamide derivatives as antioxidants and pharmaceutical compositions containing them – Google Patents
- Saritha, K.; Sarala, D.T.; Vidya, R.M.; Sudheer, K.G.; Umamaheswari, A.; Rajitha, G. Drug. Des. Discov. 2024,21(13), 2711-2727, https://doi.org/10.2174/1570180820666230816091339.
CrossRef - Sangshetti, J.; Pathan, S.K.; Patil, R.; Akber Ansari, S.; Chhajed, S.; Arote, R.; Shinde, D.B. Med. Chem.2019, 27(18), 3979-3997, https://doi.org/10.1016/j.bmc.2019.07.050.
CrossRef - El-Helby, A.A.; Sakr. H.; Ayyad, R.R.A.; El-Adl. K.; Ali, M.M.; Khedr, F. Med Chem.2018, 18(8), 1184-1196, https://doi.org/10.2174/1871520618666180412123833.
CrossRef - Go, K.; Tsurumi, K.; Fujimura, H. J. Pharmacol. 1978,28(1), 93-104, https://doi.org/10.1254/jjp.28.93.
CrossRef - Akashi, A.; Chiba, T.; Kasahara, A. J. Pharmacol. 1974, 29(1), 161-4, https://doi.org/10.1016/0014-2999(74)90183-6.
CrossRef - Sridhara, A.M.; Reddy, K.R.; Keshavayya, J.; Goud, P.S.; Somashekar, B.C.; Bose, P.; Peethambar, S.K.; Gaddam, S.K. J. Med. Chem.2010, 45(11), 4983-4989, https://doi.org/10.1016/j.ejmech.2010.08.005.
CrossRef - Awantu, A.; Fongang, Y.; Ayimele, G.; Nantia, E.; Fokou, P.; Boyom, F.; Ngwang, C.; Lenta, B.; Ngouela, S. J. Org. Chem.2020, 10, 1-16, https://doi.org/10.4236/ijoc.2020.101001.
CrossRef - Ruiz-Magaña, M. J.; Martínez-Aguilar, R.; Lucendo, E.; Campillo-Davo, D.; Schulze-Osthoff, K.; Ruiz-Ruiz, C. Oncotarget. 2016, 7(16), 21875-86, https://doi.org/10.18632/oncotarget.7871.
CrossRef - Stefany Aires do Nascimento, F. B.; do Amaral Valente Sá, L. G.; de Andrade Neto, J. B.; da Silva, L. J.; Rodrigues, D. S.; de Farias Cabral, V. P.; Barbosa, A. D.; Almeida Moreira, L. E.; Braga Vasconcelos, C. R.; Cavalcanti, B. C.; França Rios, M. E.; Silva, J.; Marinho, E. S.; Dos Santos, H. S.; de Mesquita, J. R.; Pinto Lobo, M. D.; de Moraes, M. O.; Nobre Júnior, H. V.; da Silva, C. R. Future Microbiol. 2024, 19, 91-106, https://doi.org/10.2217/fmb-2023-0160.
CrossRef - Pereira, A.M.G.; da Silva, B.F.; Araujo, I.M.F.; Aguiar, F.K.C.; Coelho, P.A.T.; Costa, R.A.; Marinho, M.M.; Marinho, E.S.; Nunes, J.V.S.; Carneiro, V.A.; Santos, H.S. Antibiotics. 2025, 14(3), 286, https://doi.org/10.3390/antibiotics14030286.
CrossRef - Chhunchha, B.; Kubo, E.; Krueger, R.R.; Singh, D.P. Antioxidants (Basel). 2023,12(1), 140, doi: https://doi.org/10.3390/antiox12010140.
CrossRef - SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Rep.2017, 7, 42717, http://www.swissadme.ch/.
CrossRef - P.; Eckert, O.A.; Schrey, A.K.; Preissner, R. ProTox-II: a webserver for the prediction of toxicity of chemicals, Nucleic Acids Research, 2018, 46, 257–263, https://doi.org/10.1093/nar/gky318.
CrossRef - Soujanya, M.; Rajitha, G.; Umamaheswari, A.; Kumar, K. S. Drug Des. Disc.2017, 15(8), 875–886, https://doi.org/10.2174/1570180814666171026161041.
CrossRef - Rajitha, G.; Vidya Rani, M.; Vankadothnaik, U.; Umamaheswari,A. J Pharm Res Int.2021, 33(46A), 470-483, https://doi.org/10.9734/jpri/2021/v33i46A32890.
CrossRef - Anowar Hosen, M.; Sultana Munia, N.; Al-Ghorbani, M.; Baashen, M.; Almalki F.A.; Ben Hadda, T.; Ali, F.; Mahmud, S.; Abu Saleh, M.; Laaroussi, H.; Kawsar, S.M.A. Chem.2022, 125, 105850, https://doi.org/10.1016/j.bioorg.2022.105850.
CrossRef - Horishny, V.; Geronikaki, A.; Kartsev, V.; Matiychuk, V.; Petrou, A.; Pogodin, P.; Poroikov, V.; Papadopoulou, T. A.; Vizirianakis, I. S.; Kostic, M.; Ivanov, M.; Sokovic, M. Molecules, 2022, 27(3), 1068, https://doi.org/10.3390/molecules27031068.
CrossRef - Margerrison, E.E.; Hopewell, R.; Fisher, LM. J Bacteriol. 1992,174(5), 1596-603, https://doi.org/10.1128/jb.174.5.1596-1603.1992.
CrossRef - Benson, T. E.; Walsh, C. T.; Hogle, J. M. Structure (London, England: 1993),1996, 4(1), 47–54. https://doi.org/10.1016/s0969-2126(96)00008-1.
CrossRef
Accepted on: 31 Dec 2025
Second Review by: Dr Arifa Begum SK
Final Approval by: Dr. Tanay Pramanik







ISSN Online: 2231-5039

![]()




{kind=link}