Integrated In Vitro, Ex Vivo, and In Silico Investigation of 4-Chlorochalcone-Derived Dihydropyrazole and Pyrimidine Analogues as Antioxidant and Anticancer Agents
 , K. M. Ferdousul Haque1, Muhammad Abdullah Al-Mansur2, Mohammad A. Rashid1and Mohammad Sharifur Rahman1*
, K. M. Ferdousul Haque1, Muhammad Abdullah Al-Mansur2, Mohammad A. Rashid1and Mohammad Sharifur Rahman1*1Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Dhaka, Dhaka, Bangladesh
2Institute of National Analytical Research and Service (INARS), Bangladesh Council of Scientific and Industrial Research (BCSIR), Dr.Qudrat-I-Khuda Road, Dhanmondi, Dhaka, Bangladesh.
Corresponding Author E-mail:msr@du.ac.bd
DOI : http://dx.doi.org/10.13005/ojc/420229
Download this article as:
![]()
Cancer and disorders associated with oxidative stress continue to pose serious health burdens worldwide due to drug resistance, side effects, and limited therapeutic efficacy. Chalcones, versatile 1,3-diaryl-2-propen-1-one compounds, are attractive scaffolds because of their broad biological activities and ability to form bioactive heterocycles. In the current investigation, the (E)-3-(4-chlorophenyl)-1-phenylprop-2-en-1-one (1) chalcone was synthesized and converted into a dihydropyrazole, 5-(4-chlorophenyl)-1,3-diphenyl-4,5-dihydro-1H-pyrazole (2), and a pyrimidine, 4-(4-chlorophenyl)-6-phenylpyrimidin-2-amine (3). All were assessed for antioxidant activity (DPPH free radical scavenging test) and anticancer effects against HeLa cells. Molecular docking with human telomerase reverse transcriptase (TERT) showed strong binding strength (−9.0 to −9.9 kcal/mol), supported by stable 100 ns molecular dynamics simulations, particularly for compound (2). Computational studies revealed distinct electronic and thermodynamic properties, with compound (2) showing high stability and reactivity. Overall, heterocyclic modification enhanced biological activity, highlighting compound (2) as a promising multifunctional anticancer candidate.
KEYWORDS:Antioxidant potential; 4-Chlorochalcone derivatives; HeLa cytotoxicity, Dihydropyrazole; Molecular dynamics simulation; Telomerase inhibition
Introduction
Chalcones are carbonyl compounds that are α,β-unsaturated and consist of two aromatic or substituted aromatic rings connected by a three-carbon enone system. The various biological actions of these compounds, such as their antiinflammatory, anti-tubercular, anticancer, and antibacterial properties, have garnered a lot of attention.1 Incorporation of electron-withdrawing elements such as chloro group may further influence lipophilicity and receptor binding affinity, thereby improving pharmacological performance. Structural modification of the chalcone backbone through heterocyclic ring formation is known to enhance molecular stability, target selectivity, and biological potency. So chalcones are useful synthetic intermediates that are used to create biologically significant heterocycles such pyrazolines, benzothiazepines, pyrimidines, and flavones.2 Dihydropyrazole derivatives are reported to exhibit antibacterial, anticancer, and anti-inflammatory activities3, whereas pyrimidine derivatives own a wide-ranging pharmacological properties, comprising antitumor, antidiabetic, anti-HIV, antimicrobial, antileishmanial, and anticancer actions.4,5
Oxidative stress may arise during routine metabolic activities, resulting in cellular injury and diminished immune protection.6,7 Pyrazole and pyrimidine derivatives have demonstrated notable antioxidant potential in previous studie.3,8 Cancer remains a major global health burden, and the limitations of current chemotherapeutic agents, such as toxicity, resistance, and high treatment costs, emphasize the need for novel anticancer compounds. Pyrazole and pyrimidine scaffolds are considered promising frameworks for anticancer drug development.3,9,10
In cancer cells, telomerase reverse transcriptase (TERT) is frequently overexpressed, enabling unlimited cell proliferation and tumor progression.11-13 Pyrazole and pyrimidine derivatives have been reported to interact with TERT.14 In silico docking analyses offer detailed atomic-level understanding of ligand–receptor interactions, thereby supporting the observed experimental biological results.Density functional theory (DFT) exploration helps to elucidate the electronic nature, reactivity, and stability of the synthesized compounds. The combined experimental and in silico approach adopted here contributes to a deeper understanding of structure–activity interactions and wires the rational design of potent active molecules. Based on these findings, the present study describes the synthesis of 4-chlorochalcone (1), its transformation into pyrazole (2), and pyrimidine (3) derivatives, followed by antioxidant, anticancer, molecular docking, and DFT studies.
Experimental Details
General
A Gallenkamp capillary instrument was employed to measure the melting points. On a Shimadzu Corporation spectrophotometer, infrared (FTIR) spectra were captured in order to pinpoint distinctive functional groups. Bruker Corporation’s 600 MHz and 150 MHz device was used to obtain proton and carbon nuclear magnetic resonance spectra. For chromatography, silica gel was used as the immobile phase for reaction monitoring plus purification. Merck Group and LobaChemie were the suppliers of all solvents, reagents, and starting materials, which were used exactly as supplied and without any additional purification.
Chalcone (1) synthesis
An equimolar (0.01 mol) ethanolic solution of acetophenone and 4-chlorobenzaldehyde were mixed with acqueous sodium hydroxide in ambient conditions. A pale yellow precipitate was formed and after the reaction mix was agitated at room temperature till it was finished. The desired component was obtained by filtering the solid product, thoroughly washing it with distilled water, and then drying it.
92% yield; pale yellow mass; m.p 114 -116 °C; FTIR (cm⁻¹): 3055, 1655, 1589, 1565, 1486, 1446–1402, 1216, and 822.1H NMR (600 MHz using CDCl3) δ 7.39 (2H, d, J = 8.40 Hz, H-6, 8), 7.50 (3H, m, H-2, 11, 13), 7.57 (3H, m, H-5, 9, 12), 7.76 (1H, d, J=15.6 Hz, H-3), 8.01 (2H, m, H-10, 14); 13C NMR (150 MHz using CDCl3): δ 190.37, 143.45, 138.16, 136.56, 133.07, 129.72, 129.38, 128.81, 128.63, 122.61.
Pyrazole (2) synthesis
Chalcone (1) and phenylhydrazine (0.01 mole each) were mixed with ethanol and heated under reflux at 80 °C for six hours. Following solvent removal, n-hexane–ethyl acetate was used as the eluent in silica gel column chromatography to purify the crude substance.
Yield 60%; brown solid; m.p. 146-148 °C; FTIR (cm⁻¹): 3062, 1595, 1500–1447, 1333, 821 ; 1H NMR (600 MHz using CDCl3) δ 3.12, (1H, dd, J = 17.2, 7.2 Hz, H-4a), 3.84 (1H, dd, J =17.2, 12.4 Hz, H-4b), 5.25 (1H, m, H-5), 7.04-7.41 (14 aromatic H, m); 13C NMR (150 MHz using CDCl3): δ 152.21, 143.39, 141.22, 134.60, 132.88, 130.11, 129.22, 128.89, 127.48, 125.52, 119.51, 113.46, 64.03, 43.63.
Pyrimidine (3) synthesis
An equimolar (0.005 mole) chalcone(1) and guanidine hydrochloride were refluxed in ethanol with 40% NaOH at 80 °C for 6 hours, followed by silica gel column chromatography purification whether n-hexane–ethyl acetate was used as the eluent.
Yield 61%, Brown solid, m.p. 149-151 ºC; FTIR (cm-1): 3493, 2922, 1580, 1357, 811;1H NMR (600 MHz using CDCl3) δ 5.41 (2H, s, NH2-2), 7.42 (1H, s, H-5), 7.46-7.52 (5 aromatic H, m), 8.01-8.06 (4 aromatic H, m); 13C NMR (150 MHz using CDCl3): δ 166.10, 164.94, 162.69, 137.62, 137.62, 136.69, 135.38, 131.24, 129.27, 129.09, 129.71, 127.40, 103.97.
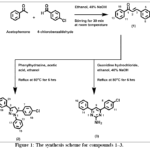 |
Figure 1: The synthesis scheme for compounds 1–3. Click here to View Figure |
Antiproliferative assay
HeLa cells were grown in 96-well plate (2.0 × 10⁴ cells per well) after being maintained in DMEM supplemented with the proper additives. 25 µL of the test chemical solution (1 mg/mL made in 1% DMSO) was added to the cells after they had been nurtured for 24 hours (at 37°C and 5% carbon dioxide). The cytotoxic effects were checked by inverted light microscopy after an additional 24 hours of treatment. Morphological fluctuations such as cell rounding, size reduction, and loss of adherence were recorded as indicators of cytotoxic response. To guarantee reproducibility, every experiment was carried out in triplicate.15,16
Free Radical Scavenging Assay by DPPH
The antioxidant test was done using the DPPH assay.17 Various concentrations of test compounds in methanol were reacted with DPPH solution for 20 minutes in darkness. Scavenging activity was determined spectrophotometrically at 517 nm, with percent inhibition calculated relative to a control. Ascorbic acid served as the standard reference.
Molecular Docking
The TERT protein (PDB: 5CQG) was prepared by removing water, ligands, and chain B, followed by adding polar hydrogens in PyMOL.18 Ligands (1–3 and BIBR1532) were sourced from PubChem and converted via GaussView.19 Energy minimization of the ligands was performed prior to docking to ensure stable and biologically relevant conformations. PyRx was used for docking within a specific grid box.Key noncovalent interactions, including H-bondings, hydrophobic connections, and π–π assemblies, were evaluated to understand ligand–protein binding stability. Optimal poses were selected by binding affinity and analyzed for residues using Discovery Studio and LigPlot+.20,21 Comparative analysis of binding energies and interaction patterns was carried out to correlate docking results with observed biological activities.
Molecular Dynamics Simulation
100 ns MD simulations were run using Desmond v6.0 to check the fixity of the TERT–ligand complexes. The TERT structure (PDB ID: 5CQG) was refined, and the ligands—chalcone (1), dihydropyrazole (2), and pyrimidine (3)—along with the standard inhibitor BIBR1532, were prepared using the OPLS3e force field.22,23 In a TIP3P water box, each complex was solvated and equilibrated under conditions of NPT at 300 K and 1.013 bar. Trajectories were analyzed for RMSD, radius of gyration, SASA, and hydrogen bonding. Results were compared against the apo protein and BIBR1532 controls.24
In Silico Drug-Like Properties
Physicochemical parameters, pharmacokinetic characteristics, drug-likeness, and medicinal chemistry filters were projected by SwissADME (http://www.swissadme.ch/).25
Study of density function theory
Structures of chalcone (1), dihydropyrazole (2), and pyrimidine (3) were obtained from PubChem and optimized using Gaussian 9.5 at the B3LYP/6 -31G++ (d,p) level.26 Vibrational frequencies confirmed stable minima with no imaginary frequencies. Merz–Kollman ESP charges and population analyses were conducted to determine electron distribution. To assess each synthesized compound’s stability and reactivity, key electronic characteristics such as HOMO-LUMO energies, and chemical hardness or softness were calculated.27
Result and Discussion
Compound 1‘s crystalline powder had a white to yellow hue. TLC analysis on silica gel PF254 after being sprayed with the vanillin-sulfuric acid reagent and heated to 90 °C showed a yellow tint and a dark area when exposed to UV light (254 nm). The material dissolved in chloroform, methanol, and ethyl acetate. Infrared spectrum absorption bands were seen at 3055 (Csp²–H), 1655 (C=O), 1589 (C=C), 1565 and 1486 (aromatic C=C), 1402–1446 (C–C), 1216 (C–O), and 822 cm⁻¹ (C–Cl) [28]. The ¹H NMR at 600 MHz using CDCl3 revealed a downfield multiplet located at δ 8.01 for H-10 and H-14. Other multiplets were appeared at δ 7.50 (H-2, H-11 & H-13) and δ 7.57 (H-5, H-9 & H-12) in addition to a doublet seen at δ 7.39 (J = 8.40 Hz) for H-6 and H-8. This was caused by the carbonyl group at C-1. In the α,β-unsaturated system, a doublet at δ 7.76 (J = 15.6 Hz) confirmed the E-configuration of H-3. The spectrum data supported the description of the chalcone (E)-3-(4-chlorophenyl)-1-phenylprop-2-en-1-one [26–28]. Further evidence for the structure came from the ¹³C NMR spectrum, which matched values found in the literature.28-30
Compound 2’s crystalline powder had a brown solid appearance. TLC on silica gel PF254 exposed a dark UV-active spot at 254 nm, which turned yellow in the presence of vanillin-sulfuric acid reagent and heating. It was found soluble in chloroform, ethyl acetate, methanol, ethanol, and other common organic solvents. The IR spectrum displayed characteristic absorptions at 3062 (Csp²–H), 1333 (C–N), 1595 (aromatic and C=C), and 821 cm⁻¹ (C–Cl) [31]. The ¹H NMR spectrum (600 MHz, CDCl₃) exhibited diagnostic signals for a 4,5-dihydro-1H-pyrazole (pyrazoline) ring system. The two diastereotopic protons at C-4 appeared as double doublets, i.e. H-4a at δ 3.12 (J = 17.2, 7.2 Hz) and H-4b at δ 3.84 (J = 17.2, 12.4 Hz). The methine proton at C-5 resonated as a multiplet at δ 5.25. A series of overlapping multiplets in the region δ 6.80–7.78 accounted for the fourteen aromatic protons. These features were found aligned with those reported in the literature.31 confirming the identity of compound 2 as 5-(4-chlorophenyl)-1,3-diphenyl-4,5-dihydro-1H-pyrazole. The ¹³C NMR spectrum further supported this assignment and was in agreement with previously published data.31
The compound 3 was found to be a brown solid. After being sprayed with the vanillin-sulfuric acid reagent and heated, TLC on silica gel PF254 became yellow and showed a dark area when exposed to UV light (254 nm). The substance showed solubility in ethyl acetate, chloroform, and ethanol. Typical absorptions were seen at 3493 (N–H stretching), 2922 (C–H), 1580 (C=C), 1357 (C–N), and 811 cm⁻¹ (C–Cl) in the infrared spectrum [32]. The ¹H NMR spectrum (600 MHz, CDCl₃) provided key structural insights. A singlet at δ 5.41 integrating for a pair of protons corresponded to the amine functional group (–NH₂) attached to C-2 of the pyrimidine moiety. Another single peak at δ 7.42 was assigned to the lone proton at C-5 of the pyrimidine nucleus. The aromatic protons of the acetophenone-derived phenyl ring (H-13 to H-18) resonated as a cluster between δ 7.46–7.52. In contrast, the aromatic protons originating from the 4-chlorobenzaldehyde moiety (H-8, H-9, H-11, H-12) appeared downfield at δ 8.01–8.06, likely because of the electron-pulling effect of the adjacent pyrimidine ring. These features were consistent with those reported in the literature [30–32], confirming the structure of compound 3 as 4-(4-chlorophenyl)-6-phenylpyrimidin-2-amine. The ¹³C NMR spectrum further supported this assignment and was in agreement with published data.32-34
The produced compounds showed moderate antioxidant activity in the DPPH radical scavenging experiment, with IC50 values 45 to 80 μg/mL (Table 1).
Compound 3 has the highest antioxidant potential, followed by Compound 2 and 1.
This suggests that the pyrimidine derivative (compound 3) possesses superior radical scavenging potential compared to the chalcone and pyrazoline analogues.
Table 1: Antioxidant and antiproliferative activities of the synthetic compounds1-3.
|
Test compounds |
DPPH assay | Antiproliferative assay using HeLa cell | |
| IC50 (μg/ml) | Survival of HeLa cells |
Remarks |
|
|
1 |
80.2 ± 0.15 | <5% | Cytotoxic |
| 2 | 47.1 ± 0.54 | <5% |
Cytotoxic |
|
3 |
45.5 ± 1.69 | <5% | Cytotoxic |
| Butylatedhydroxy toluene | 10.7 ± 0.33 | – |
– |
|
Culture media + Vehicle |
– | >95% |
Not cytotoxic |
Molecular docking revealed that compounds 1–3 and the reference TERT inhibitor BIBR1532 [35] exhibited strong binding affinities toward the TERT active site, with docking scores of –9.0, –9.6, –9.9, and –10.6 kcal/mol, respectively. Met482 (methionine), Met483 (methionine), Phe494 (phenylalanine), Gly495 (glycine), Trp498 (tryptophan), Ile550 (isoleucine), Tyr551 (tyrosine), Leu554 (leucine), and Arg557 (arginine) were identified as key amino acid residues involved in stabilizing the ligand through significant binding interactions.(Fig. 2; Table 2).
Compound 1 showed hydrophobic interactions similar to BIBR1532 with an additional Gly553 contact. Compound 2 formed an extra interaction with Asn492, while compound 3 established a stabilizing hydrogen bond with Ile550 via its –NH₂ group.
Table 2: Binding affinities and noncovalent (hydrogen bonding and hydrophobic) interactions of 1-3 with the reference inhibitor, BIBR1532.
|
Ligands |
Binding strength (Kcal/mol) |
Interactions (Three letter amino acid code) |
|
BIBR1532 |
-10.6 | Phe 494, Gly 495, Tyr 551, Leu 554, Arg 557, Ile 550, Trp 498, Met 482, Met 483 |
| 1 | -9.0 |
Arg 557, Gly 495, Gly 553, Ile 497, Ile 550, Leu 554, Met 482, Met 483, Phe 494, Trp 498, Tyr 551 |
|
2 |
-9.6 | Ile 550, Leu 554, Met 482, Met 483, Asn 492, Phe 494, Arg 557, Tyr 551, Trp 498. |
| 3 | -9.9 |
Met 482, Met 483, Phe 494, Trp 498, Ile 550 (H-bond), Tyr 551, Leu 554, Arg 557 |
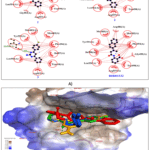 |
Figure 2: A) Noncovalent (hydrogen bonding and hydrophobic) interactions of the test compounds 1-3 and the reference inhibitor (BIBR1532) in 2D plot. B) 3D superimposed position of 1-3 & BIBR1532 in the binding pocket of TERT, where 1, 2, 3, and BIBR1532 are presented in green, yellow, blue and red color. |
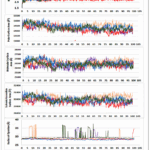 |
Figure 3: Molecular dynamics simulation studies of compounds 1-3 with standard and apoenzyme Click here to View Figure |
MD simulations (Fig. 3, Table 3) were executed to inspect solidity and dynamic manners of human telomerase reverse transcriptase (TERT, PDB ID: 5CQG) in complex with the designed ligands (compounds 1–3) and the reference inhibitor BIBR1532 [35]. RMSD analysis revealed that the apo protein was comparatively flexible, with an average RMSD of 3.53 Å, whereas the BIBR1532–TERT complex showed enhanced stability (2.88 Å). Among the synthesized compounds, compound 2 (dihydropyrazole) demonstrated the uppermost constancy with an average RMSD of 3.21 Å, followed by compound 1 (chalcone, 3.35 Å) and compound 3 (pyrimidine, 3.40 Å). Initial fluctuations during the first 10–15 ns were attributed to equilibration, after which all systems attained stable conformations comparable to the reference inhibitor.
The radius of gyration (Rg) remained stable throughout the simulations, indicating preserved structural compactness. Compound 2 exhibited the lowest average Rg value (28.32 Å), suggesting tighter protein–ligand packing. SASA values were consistent for all complexes (29,600–30,000 Å2), with compound 2 showing slightly reduced surface exposure. PSA (420–460 Ų) and MSA (29,500–30,000 Ų) analyses further supported the superior stability of compound 2. Hydrogen bond analysis showed that compound 2 formed persistent interactions with Gln851, Asp868, and Ser921, while maintaining π–π stacking and hydrophobic contacts similar to BIBR1532.35. Overall, MD simulations highlight compound 2 as the most firm and favorable TERT inhibitor scaffold.
Table 3: Summary of molecular dynamic behaviours of the compounds 1-3
|
Parameter |
Apo | Standard (BIBR1532) | Compound 1 | Compound 2 | Compound 3 |
| RMSD (Å) | 3.53 | 2.88 | 3.35 | 3.21 |
3.40 |
|
Rg (Å) |
29.28 | 28.58 | 29.31 | 28.32 | 29.57 |
| SASA (Ų) | 29,849 | — | 29,624 | 29,950 |
29,983 |
|
PSA (Ų) |
— | — | 450 | 440 | 455 |
| MSA (Ų) | 29,849 | 29,500 | 29,850 | 29,600 |
30,000 |
|
H-Bonds |
— | 2–3 | 1–2 | 2–3 (persistent) | 1–2 |
| Key Interaction Residues | — | Asp868, Tyr905 | Gln851, Asp868 | Gln851, Asp868, Ser921, Tyr905 |
Gln851, Tyr905 |
Compounds 1–3 were evaluated for their physicochemical properties, pharmacokinetic behavior, and drug-likeness using the SwissADME web platform (Table 4). The predicted TPSA values ranged from 17.07 to 51.80 Ų, signifying favorable membrane permeability and good intestinal absorption, with an additional indication of potential blood–brain barrier penetration. The calculated lipophilicity values (logP = 4.06–4.49) reflected an appropriate balance between hydrophilicity and lipophilicity, supporting oral bioavailability. Moreover, the cytochrome P450 inhibition profiles were within acceptable limits, implying a lower risk of metabolic instability and drug–drug associations. Lipinski’s classical rule of five showed that all compounds complied with the criteria for oral drugs, except compound 2, which exhibited a single violation. Further assessment using Veber, Ghose, Egan, and Muegge filters supported the overall suitability of these molecules as potential lead compounds, warranting further experimental validation.
Table 4: Summary of drug likeness study of the compounds 1-3
|
Parameters |
Compound 1 | Compound 2 | Compound 3 |
|
A) Physicochemical Properties |
|||
|
Formula |
C15H11ClO | C21H17ClN2 | C16H12ClN3 |
| Molecular weight (MW) | 242.70 | 332.83 |
281.74 |
|
No. of Heavy atoms |
17 | 24 | 20 |
| Rotatable bond | 3 | 3 |
2 |
|
Hydrogen bond acceptor |
1 | 1 | 2 |
| Hydrogen bond Donor | 0 | 0 |
1 |
|
Molar Refractivity |
71.26 | 107.27 | 82.32 |
| Mean logP | 4.49 | 5.94 |
4.06 |
|
TPSA (Topological Polar Surface Area) |
17.07A2 | 15.60A2 |
51.80A2 |
|
B) Pharmacokinetics |
|||
| GI Absorption | High | High |
High |
|
BBB infiltration |
++ | ++ | ++ |
| CYP1A2 inhbition | ++ | ++ |
++ |
|
CYP2C9 inhbition |
++ | ++ |
— |
|
C) Drug likeliness filters |
|||
| Lipinski | ++ 0 Breach | ++ 1 Breach
MLOGP >4.15 |
++ 0 Breach |
|
(Ghose, Veber, Egan) |
++ | ++ | ++ |
| Muegge | — 1violation,
Heteroatoms <2 |
— 1violation:XLOGP >5 |
++ |
++ indicates compliance and −− indicates noncompliance. Drug-likeness was evaluated using standard filters. Lipinski criteria included MW ≤ 500, HBA ≤ 10, HBD ≤ 5, and MLOGP ≤ 4.15. Ghose rules required MW 160–480, MR 40–130, 20–70 atoms, and WLOGP −0.4 to 5.6. Egan criteria were TPSA ≤ 131.6 and WLOGP ≤ 5.88. Muegge filtering included MW 200–600, TPSA ≤ 157, XLOGP −2 to 5, HBA ≤ 10, HBD ≤ 5, and rotatable bonds ≤ 15.
Computational analysis highlighted clear electronic and thermodynamic differences among compounds 1–3(Table 5 and 6). Dipole moment calculations showed that compound 1 was the most polar (4.0491 D), followed by compound 2 (2.6234 D) and compound 3 (2.3779 D), reflecting differences in molecular symmetry and functional group orientation. Thermochemical parameters indicated that compound 2 possessed more negative internal energy, enthalpy, and Gibbs free energy values, suggesting greater thermodynamic stability than compounds 1 and 3. Frontier molecular orbital analysis revealed that compound 3exhibited the widest HOMO–LUMO gap (4.400 eV), followed by compound 1 (4.043 eV), while compound 2 showed the smallest gap (3.825 eV), consistent with its lower hardness and higher softness. Electronegativity and electrophilicity values further supported these trends. The HOMO–LUMO distributions (Fig. 4) and molecular electrostatic potential maps (Fig. 5) clearly illustrated charge distribution and reactive regions, supporting the predicted reactivity and stability patterns.
Table 5: Dipole moment and specific energies of compound 1-3.
|
Compounds |
Internal energy in Hartree unit | Free energy in Hartree unit | Enthalpy in Hartree unit | Dipole moment in Debye unit |
| 1 | -1113.457 | -1113.490 | -1113.436 |
4.0491 |
|
2 |
-1379.556 | -1379.616 | -1379.922 | 2.6234 |
| 3 | -1240.923 | -1240.981 | -1240.922 |
2.3779 |
Table 6: Frontiers molecular orbital theory calculated from compound 1-3.
|
Samples |
HOMO Energy (eV) | LUMO Energy (eV) | LUMO-HOMO Gap (eV) | Hardness, ƞ (eV) | Softness, S (eV-1) | Chemical potential, µ (eV) | Electronegativity, χ (eV) |
Electrophilicity, ɷ (eV) |
|
1 |
-6.584 | -2.541 | 4.043 | 2.021 | 0.494 | -4.563 | 4.563 | 5.15 |
| 2 | -5.021 | -1.196 | 3.825 | 1.193 | 0.838 | -3.108 | 3.108 |
2.53 |
|
3 |
-6.059 | -1.659 | 4.400 | 2.200 | 0.454 | -3.859 | 3.859 |
3.38 |
Here, eV=electron volt units of energy.
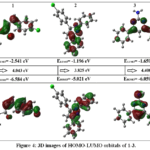 |
Figure 4: 3D images of HOMO-LUMO orbitals of 1-3. Click here to View Figure |
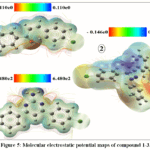 |
Figure 5: Molecular electrostatic potential maps of compound 1-3. Click here to View Figure |
Conclusion
In summary, this study successfully explored the potential of 4-chlorochalcone (1) and its known heterocyclic derivatives, dihydropyrazole (2) and pyrimidine (3). Introduction of nitrogen-containing heterocycles appears to promote stronger target engagement through improved electronic complementarity and conformational stability.Biological evaluation demonstrated that the heterocyclic derivatives exhibited markedly enhanced properties compared to the parent chalcone, showing significant free radical scavenging potential and strong antiproliferative effects across the tested models, which may be attributed to improved molecular rigidity, heteroatom incorporation, and favourable electronic distribution. Computational analyses confirmed their stable and energetically favourable interactions with the telomerase reverse transcriptase (TERT) enzyme, supporting a telomerase inhibition–based mechanism. Among the series, the dihydropyrazole derivative (2) displayed the highest structural stability and the most persistent binding mode throughout the simulation period, closely resembling the reference inhibitor BIBR1532 in both orientation and interaction pattern. Collectively, these results indicate that compound (2) represents a good framework for the developing new telomerase-targeted anticancer entities, combining potent cytotoxicity, antioxidant potential, and strong computational validation, and thus merits further optimization and advanced biological investigation.
Funding Sources
The University Grants Commission of Bangladesh awarded MSR a grant for the fiscal year 2021–2022 to support this project.
Conflict of Interest
The authors declare that there is no conflict of interest.
Authors’ Contributions
SA conducted the chemical synthesis and biological studies. KMFH performed the computational studies. MAAM carried out the NMR data acquisition and analysis. MAR critically reviewed the manuscript. MSR conceived the research theme, supervised the study, and finalized the manuscript.
Data Availability Statement
ll data supporting the findings of this study are available from the corresponding author upon reasonable request.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Abd El-Sattar, N. E. A.; Badawy, E. H. K.; Abdel-Mottaleb, M. S. A. Synthesis of Some Pyrimidine, Pyrazole, and Pyridine Derivatives and Their Reactivity Descriptors. J. Chem.2018, 8795061. https://doi.org/10.1155/2018/8795061
CrossRef - Shalaby, A. A.; El‑Sayed, N. S.; Abdel‑Rahman, H. M. Chalcones as Versatile Scaffolds in Organic Synthesis and Medicinal Chemistry: Recent Advances. Org. Biomol. Chem.2023, 21 (18), 3651–3685. https://doi.org/10.1039/D3OB00792H
CrossRef - Shaik, A. B.; Bhandare, R. R.; Nissankararao, S.; Edis, Z.; Tangirala, N. R.; Shahanaaz, S.; Rahman, M. M. Design, Facile Synthesis and Characterization of Dichloro Substituted Chalcones and Dihydropyrazole Derivatives for Their Antifungal, Antitubercular and Antiproliferative Activities. Molecules.2020, 25, 3188.https://doi.org/10.3390/molecules25143188
CrossRef - Tylińska, B.; Wiatrak, B.; Czyżnikowska, Ż.; Cieśla-Niechwiadowicz, A.; Gębarowska, E.; Janicka-Kłos, A. Novel Pyrimidine Derivatives as Potential Anticancer Agents: Synthesis, Biological Evaluation and Molecular Docking Study. Int. J. Mol. Sci.2021, 22, 3825.https://doi.org/10.3390/ijms22083825
CrossRef - Rathika, G.; Mythili, S.; Naveentha, X. P.; et al. Exploring the Anticancer Potential of Pyrimidine Derivatives via Molecular Docking and Dynamics. Discover Appl. Sci.2025, 7, 1319. https://doi.org/10.1007/s42452-025-07840-2
CrossRef - Kozlov, A. V.; Javadov, S.; Sommer, N. Cellular ROS and Antioxidants: Physiological and Pathological Role. Antioxidants2024, 13 (5), 602.https://doi.org/10.3390/antiox13050602
CrossRef - Gerwyn, M.; Gevezova, M.; Sarafian, V.; Maes, M. Redox Regulation of the Immune Response. Cell. Mol. Immunol.2022, 19, 1079–1101. https://doi.org/10.1038/s41423-022-00902-0
CrossRef - Rani, J.; Saini, M.; Kumar, S.; Verma, P. K. Design, Synthesis and Biological Potentials of Novel Tetrahydroimidazo[1,2-a]pyrimidine Derivatives. Chem. Cent. J.2017, 11, 16. https://doi.org/10.1186/s13065-017-0245-9
CrossRef - Mahapatra, A.; Prasad, T.; Sharma, T. Pyrimidine: A Review on Anticancer Activity with Key Emphasis on SAR. Future J. Pharm. Sci.2021, 7, 123.https://doi.org/10.1186/s43094-021-00274-8
CrossRef - Albratty, M.; Alhazmi, H. A. Novel Pyridine and Pyrimidine Derivatives as Promising Anticancer Agents: A Review. Arab. J. Chem.2022, 15, 103846. https://doi.org/10.1016/j.arabjc.2022.103846
CrossRef - Kang, H.; Lee, J.; Kim, S.; Kim, Y.; Park, J. Telomeres, Telomerase, and Cancer: Mechanisms, Biomarkers, and Therapeutic Implications. Exp. Hematol. Oncol.2025, 14, 7.https://doi.org/10.1186/s40164-025-00597-9
CrossRef - Kim, N. W.; Piatyszek, M. A.; Prowse, K. R.; Harley, C. B.; West, M. D.; Ho, P. L.; Coviello, G. M.; Wright, W. E.; Weinrich, S. L.; Shay, J. W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science1994, 266, 2011–2015. https://doi.org/10.1126/science.7605428
CrossRef - Baginski, M.; Serbakowska, K. In Silico Design of Telomerase Inhibitors.Drug Discov. Today2020, 25, 1213–1222. https://doi.org/10.1016/j.drudis.2020.04.024
CrossRef - Jawarkar, R. D.; Bakal, R. L.; Khatale, P. N.; Lewaa, I.; Jain, C. M.; Manwar, J. V. QSAR, PharmacophoreModeling, and Molecular Docking Studies to Identify Structural Alerts for Some Nitrogen Heterocycles as Dual Inhibitors of Telomerase Reverse Transcriptase (TERT) and Human Telomeric G-Quadruplex DNA. Future J. Pharm. Sci.2021, 7, 231.https://doi.org/10.1186/s43094-021-00380-7
CrossRef - Graziani, G.; de Maria, R.; Del Bufalo, D. In Vitro Cytotoxicity Evaluation Using Human Cancer Cell Lines.J. Vis. Exp.2023, (196), e64902.https://doi.org/10.3791/64902
CrossRef - Chen, F.; Wang, H.-M.; Kong, L.-Q.; Chen, Q.-H.; Ke, L.-N.; Dai, H.-L.; Zeng, X.-H. Synthesis and Anticancer Activity of Rhopaladins’ Analog RPDPD against the HeLa Human Cervical Cancer Cell Line.Front. Chem.2022, 10, 921276.https://doi.org/10.3389/ fchem.2022.921276
CrossRef - Ahmed, F.; Rahman, M. S. Preliminary Assessment of Free Radical Scavenging, Thrombolytic and Membrane Stabilizing Capabilities of Organic Fractions of Callistemon citrinus (Curtis.) Skeels Leaves.BMC Complement.Altern.Med.2016, 16, 247.https://doi.org/10.1186/s12906-016-1239-1
CrossRef - Seeliger, D.; de Groot, B. L. Ligand Docking and Binding Site Analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des.2010, 24, 417–422. https://doi.org/10.1007/s10822-010-9352-6
CrossRef - Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, 2009.
- Saeed, M.; Shoaib, A.; Tasleem, M.; Alabdallah, N. M.; Alam, M. J.; Asmar, Z. E.; Jamal, Q. M. S.; Bardakci, F.; Alqahtani, S. S.; Ansari, I. A.; Badraoui, R. Assessment of Antidiabetic Activity of the Shikonin by Allosteric Inhibition of Protein-Tyrosine Phosphatase 1B (PTP1B) Using State of Art: An In Silico and In Vitro Tactics. Molecules2021, 26, 3996.https://doi.org/10.3390/molecules26133996
CrossRef - Laskowski, R. A.; Swindells, M. B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model.2011, 51, 2778–2786. https://doi.org/10.1021/ci200227u
CrossRef - Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J. M.; Lu, C.; Dahlgren, M. K.; Mondal, S.; Chen, W.; Wang, L.; Abel, R.; Friesner, R. A.; Harder, E. D. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput.2019, 15, 1863–1874. https://doi.org/10.1021/acs.jctc.8b01026
CrossRef - Bougueroua, S.; Kolganov, A. A.; Helain, C.; Zens, C.; Barth, D.; Pidko, E. A.; Gaigeot, M.-P. Exploiting Graph Theory in Molecular Dynamics Simulations for Extracting Chemical and Physical Properties of Materials. Phys. Chem. Chem. Phys.2025, 27, 1298–1309. https://doi.org/10.1039/D4CP02764G
CrossRef - Ahmad, S.; Raza, K. Identification of 5-Nitroindazole as a Multitargeted Inhibitor for CDK and Transferase Kinase in Lung Cancer: A Multisampling Algorithm-Based Structural Study. Mol. Divers.2024, 28, 1189–1202. https://doi.org/10.1007/s11030-023-10648-0
CrossRef - Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep.2017, 7, 42717.https://doi.org/10.1038/srep42717
CrossRef - Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; . Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, 2016.
- Wang, D.; Lü, R.; Yuan, M.; Fu, A.; Chu, T. A DFT/TD-DFT Study of Thiazolidinedione Derivative in Dimethylformamide: Cooperative Roles of Hydrogen Bondings, Electronic and Vibrational Spectra. Spectrochim.ActaA Mol. Biomol. Spectrosc.2014, 125, 131–137. https://doi.org/10.1016/j.saa.2014.01.094
CrossRef - Wong, K. T.; Osman, H.; Parumasivam, T.; Supratman, U.; Che Omar, M. T.; Azmi, M. N. Synthesis, Characterization and Biological Evaluation of New 3,5-Disubstituted-Pyrazoline Derivatives as Potential Anti-Mycobacterium Tuberculosis H37Ra Compounds. Molecules2021, 26, 2081.https://doi.org/10.3390/molecules26072081
CrossRef - Ghoshir, U.; Kande, S.; Muley, G.; Gambhire, A. Synthesis and Characterization of Co-Doped Fly Ash Catalyst for Chalcone Synthesis. Asian J. Chem.2019, 31, 2165–2172. https://doi.org/10.14233/ajchem.2019.22053
CrossRef - Mameda, N.; Peraka, S.; Kodumuri, S.; Chevella, D.; Banothu, R.; Amrutham, V.; Nama, N. Synthesis of α,β-Unsaturated Ketones from Alkynes and Aldehydes over Hβ Zeolite under Solvent-Free Conditions. RSC Adv.2016, 6, 58137–58141. https://doi.org/10.1039/C6RA11593D
CrossRef - Yadav, C. S.; Krishna, A.; Singh, S. P. Synthesis, Characterization, and Bio‑Evaluation of Novel Series of Pyrazoline Derivatives as Potential Antifungal Agents. Sci. Rep.2025, 15, 14752.https://doi.org/10.1038/s41598-025-98645-1
CrossRef - Nikalje, A. P. G.; Tiwari, S. V.; Sangshetti, J. N.; Damale, M. D. Ultrasound-Mediated Synthesis, Biological Evaluation, Docking and In Vivo Acute Oral Toxicity Study of Novel Indolin-2-One Coupled Pyrimidine Derivatives. Res. Chem. Intermed.2018, 44, 3031–3059. https://doi.org/10.1007/s11164-018-3292-5
CrossRef - Kumar, N.; Drabu, S.; Shalini, K. Synthesis and Pharmacological Screening of 4,6-Substituted Di-(Phenyl) Pyrimidin-2-Amines. Arab. J. Chem.2017, 10, S877–S880. https://doi.org/10.1016/j.arabjc.2012.12.023
CrossRef - De, R.; Sengupta, U.; Savarimuthu, A.; Misra, S.; Nanda, J.; Bera, M. K. A Practical and Cost-Effective Approach to Polysubstituted Pyrimidine Derivatives via DBU Mediated Redox Isomerization of Propargyl Alcohol and Subsequent N–C–N Fragment Condensation. New J. Chem.2022, 46, 10603–10610. https://doi.org/10.1039/D2NJ00586G
CrossRef - Altamura, G.; degliUberti, B.; Galiero, G.; De Luca, G.; Power, K.; Licenziato, L.; Maiolino, P.; Borzacchiello, G. The Small Molecule BIBR1532 Exerts Potential Anti-Cancer Activities in Preclinical Models of Feline Oral Squamous Cell Carcinoma Through Inhibition of Telomerase Activity and Down-Regulation of TERT. Front. Vet. Sci.2021, 7.https://doi.org/10.3389/fvets.2020.620776
CrossRef - Deswal, L.; Verma, V.; Kumar, D.; Deswal, Y.; Kumar, A.; Kumar, R.; Parshad, M.; Bhatia, M. Synthesis, Antimicrobial and α-Glucosidase Inhibition of New Benzimidazole-1,2,3-Triazole-Indoline Derivatives: A Combined Experimental and Computational Venture. Chem. Pap.2022, 76, 7607–7622. https://doi.org/10.1007/s11696-022-02436-1
CrossRef - Haritha, M.; Sreerag, M.; Suresh, C. H. Quantifying the Hydrogen Bond Propensity of Drugs and Its Relationship with Lipinski’s Rule of Five. New J. Chem.2024, 48, 4896–4908. https://doi.org/10.1039/D3NJ05476D
CrossRef - Nhlapho, S.; Nyathi, M. H.; Ngwenya, B. L.; Dube, T.; Telukdarie, A.; Munien, I.; Vermeulen, A.; Chude‑Okonkwo, U. A. Druggability of Pharmaceutical Compounds Using Lipinski Rules with Machine Learning. Sci. Pharm.2024, 3 (4), 177–192. https://doi.org/10.58920/sciphar0304264.
CrossRef
Accepted on: 04 Apr 2026
Second Review by: Dr. Ashwini A. Waoo
Final Approval by: Dr. B. K Sharma







ISSN Online: 2231-5039

![]()




{kind=link}