Design of New Curcumin Derivatives with Improved Antiprostatic Activity
 , Doh Soro2and Kafoumba Bamba1
, Doh Soro2and Kafoumba Bamba11Laboratoire de Thermodynamique et de Physico-chimie du Milieu, UFR SFA, Université Nangui Abrogoua
2Université de Sam Pedro BP Abidjan, Côte-d’Ivoire
Corresponding Author E-mail:1997sageme@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/420209
Download this article as:
![]()
Curcumin and its derivatives constitute a class of promising cytotoxic agents in the fight against prostate cancer. However, as with all drugs, the efficacy of these cytotoxic agents could be hampered by the phenomenon of resistance, hence the need to develop new drugs with improved activity. This study falls within this context, with the overall objective of designing new derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diene-3-one with improved anti-prostate activity. This anti-cancer agent design uses QSAR models and theoretical chemistry methods to determine the descriptors needed to theoretically evaluate the anti-prostate cytotoxicity of the new compounds. As a result, this study has led to the design of eleven (11) new molecules designated by the code IM. The inhibitory concentration values of these curcumin derivatives are higher than those of the base compound (pIC50 = 6.678). Moreover, all the new compounds, except the IM11 analog, are within the applicability domain of the RQSA model employed. These compounds comply with Lipinski's rule. In addition, the pharmacokinetic properties determined revealed that new molecules can be used as drugs against prostate cancer. However, IM4 stands out as the most promising candidate. Not only does it exhibit improved anti-prostatic activity and an excellent ADME profile, but it also poses no risk of mutagenicity or carcinogenicity in predictive models.
KEYWORDS:ADMET; Anti-prostatic activity; Design; (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diene-3-one; Lipinski's rule
Introduction
Cancer encompasses a wide range of pathological conditions marked by uncontrolled cellular proliferation and the capacity for local invasion and distant metastasis. It arises in almost any organ or tissue and develops through a complex, multistage progression from premalignant lesions to malignant neoplasms.1 Historically viewed as a “disease of the rich,” Cancer remains one of the primary causes of death worldwide. In 2020, according to the World Health Organization (WHO) that, nearly a total of There were an estimated 19.3 million new cancer cases worldwide, accompanied by 10 million fatalities attributed to this disease2 reporting in top four cancerswere lung (11.4%), colorectal (10%), prostate (7.3%), and stomach (5.6%)).3,4 Sub-Saharan Africa is one of the most affected areas in Africa, with an increase in cancer morbidity.5 Prostate cancer occurs in the average age group of 63 years old in a metastatic state in 80% of cases. This type of cancer killed nearly 1,600 people in 2020, representing a mortality rate of 29.5% per 100,000 men.[4]. Mortality rates will keep growing because delayed symptoms often lead to late-stage detection.6 Management strategies for this cancer include surgery, radiotherapy, chemotherapy, targeted agents, hormonal therapy, and immunotherapy. Due to delayed diagnosis and advanced disease at presentation, androgen deprivation therapy and chemotherapy are most frequently used. As a systemic treatment, chemotherapy is effective in targeting cancer cells that have spread beyond the primary prostatic site.7 Chemotherapy contributes to tumor debulking prior to surgery, thereby improving surgical feasibility and potentially patient prognosis. It is primarily indicated for advanced or metastatic prostate cancer and is commonly used in combination regimens. Frequently administered agents include docetaxel, mitoxantrone, and cabazitaxel.8 Apart from side effects (nausea, loss of libido, erectile dysfunction, and weight fluctuations), over time, drugs become less effective due to the resistance that cancer cells can develop. Therefore, it is imperative to develop new anticancer agents with improved activity to anticipate these resistance phenomena. The plant kingdom offers a range of plant species with anti-prostatic properties. Turmeric is a species of perennial rhizomatous herbaceous plant of the genus Curcuma, native to South and Southeast Asia. The yellow spice of the same name is extracted from its rhizomes, which are ground into powder. Turmeric, particularly its active compound curcumin, has attracted growing interest in light of its potential properties as a therapeutic approach in cancer, including prostate cancer.9 Curcumin exhibits strong anti-inflammatory effects, which may help mitigate inflammation associated with tumor cell proliferation. It serves as an antioxidant, helping to shield cells from free radical–induced damage, and this could help prevent the development of cancer. In addition, the effectiveness of the role of curcumin in prostate cancer treatment has been increasingly explored studied in vivo and in vitro.10 Curcumin derivatives were also synthesized and tested on prostate cancer strains by Rubing Wang et al.11 These analogues of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-dien-3-one lead to the death of prostate cancer cells. In order to elucidate the origin of the biological properties of these compounds, we established a quantitative structure-activity relationship for anti-prostate activity in a previous study.12 This QSAR study focused on a series of seventeen derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-dien-3-one. The QSAR computational model was obtained employing multiple linear regression (MLR) utilized in the XLSAT software:
pIC50théo= 18,78652-4,25231* η -0,03003*α(C-C=C) -0,03074*TSurface-0,25950*Densité
This model relates the inhibition potential (pIC50) to four descriptors, namely hardness (η), bond angle α(C-C=C), surface tension (TSurface), and density. Hardness and bond angle are calculated after geometry structural optimization and vibrational analysis using B3LYP/6-31+G(d,p) theory.12 The surface tension and density descriptors were obtained using ChemSketch software. Hardness (η) proved to be the key descriptor in predicting cytotoxic potential, with the model’s statistical indicators (R2=0,922 ; S= 0,068 ; F= 17,027) have demonstrated robustness and good predictive power. Internal validation using the leave-out method and internal validation based on Tropsha’s criteria and the similarity test confirmed the quality and performance of our model. In addition, Shapiro-Wilk and Durbin-Watson tests were performed. This model is not random according to the randomization test and may be applied for estimating the cytotoxic effects of curcumin derivatives that are part of the same applicability domain subsequently, the model’s applicability was determined by analyzing Williams diagram (threshold lever . This model can be used to theoretically determine the cytotoxicity of curcumin derivatives with a leverage value of less than 0.8824. The results of cross-validation and applicability domain analysis demonstrate that the model is robust and suitable for making reliable predictions on novel molecules derived from (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diene-3-one. It is in this context that the present work is situated, the overall objective of which is to design derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diene-3-one with improved cytotoxic activity in order to anticipate resistance phenomena.
Materials and Methods
Conception
In the late 1990s, fragment-based drug design (FBDD) enabled the development of drugs by combining small molecular fragments [12]. This strategy is based on screening fragments of low molecular weight organic molecules, allowing chemical diversity to be explored efficiently.13 These fragments are actually subunits of lead compounds, commonly used in high-throughput screening for drug discovery.14 To predict the cytotoxicity of a new molecule, it is necessary to ensure that it does not contain any fragments that are absent from the dataset used to develop the model. If this is the case, the estimated result is considered reliable; otherwise, it is considered unreliable.15 In our study, the basic structure used is shown in Figure 1.
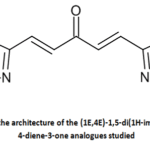 |
Figure 1: Common the architecture of the (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diene-3-one analogues studied Click here to View Figure |
Different substituents R1 and R2 are placed on the structure, and then the cytotoxic activity is evaluated.
Calculation method and molecular descriptors
All optimizations and computational analyses were performed using Gaussian 09 software.16. For the various calculations in our study, density functional theory (DFT) was used at the B3LYP/6-31+G(d,p) level.17 The descriptors used in the QSAR model are derived from overall reactivity and 3D and 2D geometries. The descriptors are chemical hardness, bond angle α(C-C=C), surface tension (TSurface), and density. Chemical hardness measures the property of a system that resists changes in its number of particles of electrons. It is expressed as follows:
η=IP – EA
In this mathematical formula, IP is the ionization potential, which is the opposite of the HOMO energy, and EA represents the electron affinity, which is the opposite of the LUMO energy. The bond angle descriptor α(C-C=C) is illustrated in Figure 2. As a fundamental property of liquids, surface tension is a key factor in many physical and chemical phenomena. Surface tension plays a crucial role in biological processes, including respiration, gas exchange in the pulmonary alveoli, and the transport of substances in blood vessels. Density helps to elucidate the behavior and characteristics of molecules in different environments.
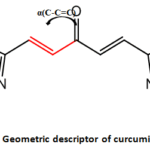 |
Figure 2: Geometric descriptor of curcumin analogues Click here to View Figure |
Lever calculation: Scope of applicability
The use of the leverage factor ensures the reliability of activity forecasts using an ARMA model.18 Leverage is a parameter that indicates the extent to which a particular piece of data contributes to or modifies the model’s forecasts. It is calculated from the model’s explanatory variable matrix. For a given observation, parameter i is determined by the following mathematical relationship:
![]()
xi is defined as the row feature vector describing compound i
X corresponds to the model matrix using descriptor values from the training set, the model is defined, where the T index indicates a transposed matrix or vector.
An RQSA model has a threshold leverage value that determines whether the model is capable of effectively predicting the activity of a given molecule. When the leverage of a molecule is below the model’s leverage threshold, the theoretical activity is acceptable; otherwise, activity prediction is impossible.
Formation thermodynamic quantities
The thermodynamic properties describing the molecules were calculated after geometric optimization of the structure. The DFT/B3LYP/6-31+G(d,p) methodology was used for all calculations related to structural analysis. For thermodynamic quantities such as the entropy of formation, the enthalpy of formation, and the free enthalpy of formation of the new anti-prostate derivatives studied were calculated using the equations presented by Otchersky and al. 19
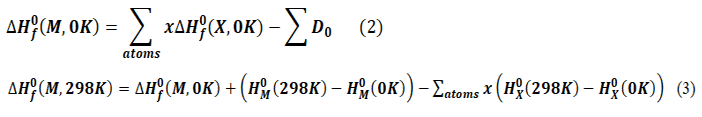
Avec :
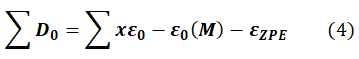
∑D0 : Atomization energy;
ε0 (M) : The total energy calculated for the molecule
εZPE: Fundamental vibrational energy of the molecular system;
HX0 (298K) – HX0 (0K) Corrections to the standard enthalpy of atomic species, provided in the JANAF database.20
HM0 (298K) – HM0 (0K) = Hcorr – εZPE (M) : Molecular Enthalpy Correction
Hcorr: Thermal correction to the enthalpy was applied.
x: Count for each atom in the molecule of type X in the molecular entity
![]()
The Rule of Five (Lipinski’s rules)
The rules proposed by Lipinski make it possible to estimate the oral absorption efficiency of a compound based on the flat structure of the molecule. The standards applicable to physicochemical characteristics were defined following a study of 2,245 drugs, both those available on the market and those currently being synthesized.21
The compound’s molecular mass should not exceed 500 g/mol;
Number of constituent atoms capable of accepting hydrogen bonds must not exceed 10;
Maximum of 5 hydrogen bond donors are allowed;
The M LogP parameter must remain below or equal to 5.
Chemical entities that do not meet minimum of two concerning the physicochemical requirements are very inclined to have absorption issues. The listed criteria establish a link between physicochemical activities and oral administration. A molecule that does not meet at least two of these criteria may show reduced absorption when administered orally.22
In silico prediction of ADMET profiles was carried out for all compounds.
HIA represents the human intestinal capacity to absorb a drug, with absorption considered low for values between 0 and 20%, medium for values between 20 and 70%, and high for values between 70 and 100%.
Permeability across Caco-2 and MDCK cells was evaluated (nm/s) measurements are used to assess the compound’s permeability in the human intestine, where a value below 4 nm/s indicates poor permeability, a value values lying between 4 and 70 nm/s indicates average permeation capacity, and a value exceeding 70 nm/s indicates high permeability for Caco-2 and MDCK
BBB is an indicator of the compound’s capability for central nervous system entry, which acts as a regulator for the passage of blood substances into the Central Nervous System.
PBB indicates the level of attachment of drugs to plasma proteins, with low binding for values below 90% and high binding for values above 90%.
CYP enzymes are essential for the metabolism of various compounds from different molecules, whether they originate inside or outside the body. They exist in several isoforms (CYP1A2, CYP2C19, CYP2D6, CYP3A4), among which the most influential are CYP2D6 and CYP3A4. It was necessary to evaluate the binding patterns of our most effective inhibitors with these isoenzymes, as their suppression can contribute significantly to drug interactions associated with toxicity or adverse effects.23
The hERG is a gene responsible for the production of a voltage-dependent potassium channel, regulating the removal of potassium from the cell. Inhibition of this channel can lead to cardiac fibrillation, which can sometimes cause failure of cardiac function.
The AMES test is a technique used to assess the compound’s mutagenicity profile.24 It makes use of various Salmonella typhimurium strains that have undergone variations in gene sequences essential necessary for histidine synthesis, making them dependent on histidine for growth. This test determines whether a compound can cause a mutation that allows the bacteria to these compounds can grow on a medium without histidine, based on values obtained from the PreADMET online tool.25
Results and Discussion
Molecular architecture of newly designed derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diène-3-one
Based on the basic structure shown in Figure 1, substituents R1 and R2 are added taking into account the shape and nature of the substituents, leading to the proposal of eleven (11) innovative molecular structures. The coded structures (IM) data obtained can be found in Table 1. Based on the optimized geometry at the B3LYP/6-31+G(d,p) level and the 2D structures concerning the compounds, the chemical hardness, bond angle α(C-C=C), surface tension TSurface, and density descriptors are determined. The recorded data for these descriptors are noted in Table 2. These descriptors are used to determine the inhibition potential of the designed analogues.
Analysis of Table 3 reveals the theoretical inhibition potential of the eleven (11) new molecules varies from 6.686à 6.955. These values of the of these new molecules are superior to those of the compounds that were used to establish the model (pIC50 vary from 6.469 à 6.678). It follows that all new derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diène-3-one have anti-prostatic cytotoxic activity (IC50) higher than those composed of the experimental database. They are therefore more effective for treating prostate cancer. The hii leverage values of the different molecules are also compared using the diagram in Figure 3.
 |
Table 1: Molecular structures of new derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diène-3-one. Click here to View Table |
Table 2: The model descriptor values along with the predicted inhibitory concentrations are presented in terms of pIC50.
|
CODES |
η | α(C-C=C) | TSurface | Densité | pIC50 | hii |
| IM1 | 1.55 | 120.695 | 50.100 | 1.260 | 6.703 |
0.795 |
|
IM2 |
1.559 | 120.749 | 46.400 | 1.190 | 6.795 | 0.091 |
| IM3 | 1.567 | 120.759 | 48.300 | 1.220 | 6.695 |
0.404 |
|
IM4 |
1.563 | 120.768 | 46.600 | 1.190 | 6.774 | 0.145 |
|
IM5 |
1.554 | 120.823 | 47.300 | 1.200 | 6.786 | 0.221 |
| IM6 | 1.568 | 120.747 | 42.800 | 1.150 | 6.879 |
0.535 |
|
IM7 |
1.556 | 120.741 | 48.300 | 1.220 | 6.743 | 0.141 |
| IM8 | 1.555 | 120.792 | 49.400 | 1.220 | 6.711 |
0.316 |
|
IM9 |
1.548 | 120.813 | 46.900 | 1.190 | 6.827 | 0.505 |
| IM10 | 1.561 | 120.768 | 49.400 | 1.220 | 6.686 |
0.422 |
|
IM11 |
1.554 | 120.412 | 43.500 | 1.140 | 6.931 | 0.961 |
 |
Figure 3: Diagram illustrating the leverage levels for the new compounds relative to the threshold leverage Click here to View Figure |
All molecules except molecule 11 are part of the same domain of validity of the QSAR model used. The QSAR model is therefore appropriate for estimating the inhibition potential of these 10 compounds. However, the leverage of the IM11 molecule (hii = 0,961)Since it is exceeding the model’s leverage threshold (h* = 0.882), the predicted its anti-prostatic activity based on the model seems uncertain.
Analysis of thermodynamic quantities associated with molecule formation
Based on the formula developed by Otchersky and al, the standard thermodynamic formation quantities, namely the formation enthalpy ΔfH°, the formation entropy ΔfS°, and the formation free enthalpy ΔfG° were determined to evaluate the formation of new, more active derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-dien-3-one. It is important to note a change in enthalpy indicates the heat exchange of a chemical reaction, whereas a change in entropy indicates the degree of thermodynamic irregularity. Finally, a change as a change in free enthalpy indicates the spontaneous tendency of the process. The estimated these quantities were quantified as presented in Table 3.
Table 3: Formation thermodynamic parameters of IM derivatives optimized at the level B3LYP/6-31+G(d,p)
|
CODES |
ΔfH°(kcal/mol) | ΔfS°(kcal/mol) |
ΔfG°(kcal/mol) |
|
IM1 |
-1140.635 | -1.404 | -722.129 |
| IM2 | -1234.726 | -1.569 |
-766.790 |
|
IM3 |
-1232.922 | -1.567 | -765.844 |
| IM 4 | -1269.324 | -1.651 |
-777.010 |
|
IM5 |
-1231.284 | -1.570 | -763.062 |
| IM6 | -1253,990 | -1,538 |
-744,563 |
|
IM7 |
-1231.161 | -1.571 | -762.809 |
| IM8 | -1245.730 | -1.572 |
-777.020 |
|
IM9 |
-1089.525 | -1.295 | -703.290 |
| IM10 | -1249.788 | -1.568 |
-782.379 |
|
IM11 |
-1173.774 | -1.457 |
-739.470 |
Analysis of the thermodynamic parameters reveals that all of the compounds studied exhibit thermodynamically favorable formation. The ΔfH° values are all strongly negative (from −1089 to −1269 kcal/mol), indicating that their formation is enthalpically favorable. Among the derivatives analyzed, IM4 (−1269.324 kcal/mol) has the most negative value, reflecting maximum enthalpic stability. The compounds IM6 (−1253.990 kcal/mol) and IM10 (−1249.788 kcal/mol) also show significant stabilization. Conversely, IM9 (−1089.525 kcal/mol) has the least negative value, suggesting weaker enthalpic stabilization and a relatively less favorable thermodynamic character compared to the other compounds. Next, the ΔfS° values are also negative (from −1.295 to −1.651 kcal/mol), indicating that the formation of molecules is accompanied by a decrease in disorder, probably linked to molecular structuring or stiffening. IM4 (−1.651 kcal/mol) shows the most marked decrease in entropy, reflecting a more orderly formation, while IM9 (−1.295 kcal/mol) shows the smallest change in entropy. However, the amplitude of entropic variations remains limited compared to enthalpic variations, suggesting that the entropic effect plays a secondary role in overall stability. Finally, the ΔfG° values, which integrate enthalpy and entropy contributions, are strongly negative (from −703 to −782 kcal/mol), confirming the spontaneous nature of the formation of all compounds. IM10 (−782.379 kcal/mol) has the most negative value, corresponding to the highest overall thermodynamic stability, followed closely by IM8 (−777.020 kcal/mol) and IM4 (−777.010 kcal/mol). In contrast, IM9 (−703.290 kcal/mol) appears to be the least thermodynamically favorable compound. Overall, the negative values of ΔfH° and ΔfG° confirm the favorable and spontaneous nature of the formation of the compounds studied. Comparative analysis indicates that overall stability is mainly governed by enthalpic contributions, with entropic effects playing a secondary role.
Measurement of Lipinski’s parameters
Lipinski’s rules are a useful guide for evaluating a compound’s potential as a drug. Molecules that comply with these rules tend to be better absorbed and distributed in the body. Lipinski’s rules have been verified for the molecules listed in Table 4. The quantities associated with Lipinski’s parameters, including molar mass, number of hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), and lipophilicity (MlogP), are also found in Table 4.
Table 4: New derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-diène-3-one, characterized by Lipinski’s parameters
|
Codes |
M(g/mol) | HBD | HBA | MlogP |
| IM1 | 382.46 | 0 | 3 |
3.14 |
|
IM2 |
410.51 | 0 | 3 | 3.55 |
| IM3 | 410.51 | 0 | 3 |
3.55 |
|
IM4 |
424.54 | 0 | 3 | 3.75 |
| IM5 | 410.51 | 0 | 3 |
3.55 |
|
IM6 |
398.5 | 0 | 3 | 3.35 |
| IM7 | 410.51 | 0 | 3 |
3.55 |
|
IM8 |
410.51 | 0 | 3 | 3.55 |
| IM9 | 356.42 | 0 | 3 |
2.71 |
|
IM10 |
410.51 | 0 | 3 | 3.55 |
| IM11 | 384.47 | 0 | 3 |
3.14 |
|
Rule |
<500 | <5 | <10 |
<4.15 |
The molecular weights of the compounds range from 356.42 g/mol to 424.54 g/mol. Molecules weighing less than 500 g/mol are considered light. These molecules are therefore more easily absorbed and pass through biological membranes. These drugs are sufficiently soluble in water to pass through biological membranes. This solubility is confirmed by the low Log P partition coefficient values, which range from 2.71 to 3.75 (Log P<4.15). In addition, the compound’s capacity for of these molecules traversing membranes is not affected by the extent of hydrogen bond donation. In fact, these curcumin analogues contain no hydrogen bond donor atoms. Furthermore, regardless of the analogue, the data show that the count of hydrogen bond acceptors registers below10. This number of donors is an advantage for absorption and diffusion through the membranes of human tissue, as a high number of acceptors can reduce the lipophilicity of the compound. As all of Lipinski’s criteria are met, these compounds can be considered as drugs that can be administered orally.
Evaluation of ADMET parameters for newly designed molecules
The ADMET behavior of newly developed molecules was assessed to predict their drug-likeness are used to assess their acceptability as drugs. These properties are predicted computational predictions were conducted using the PreADMET tool. The parameters determined include Human intestinal absorption measured via Caco-2 monolayer modelcellular diffusion capacity, in vitro MDCK cell permeability, plasma protein binding, blood-brain barrier penetration, inhibition of cytochrome P450 enzymes (CYP2D6, CYP2C9, CYP2C19, CYP2A4), inhibition of hERG, as well as cancer and mutation-related toxicity assessment. Table 5 compares the drug-likeness parameters of eleven molecules (IM1 to IM11) with those of curcumin.
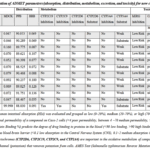 |
Table 5 : Estimation of ADMET parameters (absorption, distribution, metabolism, excretion, and toxicity) for new compounds Click here to View table |
The candidate molecules exhibit significantly higher absorption than curcumin, with HIA (human intestinal absorption) values exceeding 97%. This is a crucial advantage for improving their oral bioavailability. However, once absorbed, their distribution is similar to that of curcumin. They bind strongly protein binding in plasma and have difficulty crossing the blood-brain barrier (BBB). They would therefore not be effective in targeting brain disorders. All candidate molecules have a safer metabolic profile, with no inhibition of CYP enzymes, which reduces the risk of drug interactions. In contrast, curcumin inhibits several CYP enzymes, raising concerns when taken with other drugs. As for toxicity, the results are mixed, as curcumin is non-mutagenic and its carcinogenicity varies depending on the species (negative in mice, positive in rats). However, several candidate molecules are mutagenic (IM1, IM3, IM6, IM7, IM8, IM9, IM10) and/or carcinogenic (IM1, IM3, IM6, IM7, IM8, IM9), which is a major obstacle to their development. The IM4 molecule is the only one with a non-mutagenic and non-carcinogenic profile. Ultimately, all candidate molecules significantly improve absorption and have a better metabolic profile than curcumin, but most have toxicity issues (carcinogenicity and mutagenicity) that compromise their potential. The IM4 molecule is the most promising, as it combines better absorption and safer metabolism with a more favorable toxicity profile..
Conclusion
This study aimed to design new derivatives of (1E,4E)-1,5-di(1H-imidazol-2-yl)penta-1,4-dien-3-one, a curcumin analog structure, with a view to obtaining improved anti-prostatic activity. Using a fragment-based approach, eleven new molecules were designed. The effect of these the parameters of the compounds were quantified using an RQSA model. These anti-prostatic agents exhibit higher pIC50 inhibition potential than the compounds used to generate the model. They therefore have improved anticancer activity. These candidate molecules have a promising pharmacokinetic profile. They all comply with Lipinski’s rules, suggesting good oral bioavailability and high intestinal absorption potential. These data are confirmed by ADMET predictions, which indicate excellent human intestinal absorption (HIA > 97%) for all molecules, a significant advantage over the low bioavailability of the original curcumin. In addition, their metabolism is safer, with no inhibition of cytochrome P450 enzymes, which reduces the risk of drug interactions.
However, the study reveals major concerns regarding the toxicity of most of these derivatives. Their carcinogenicity and mutagenicity profiles, particularly for molecules such as IM1, IM3, IM6, IM7, IM8, and IM9, pose a serious obstacle to their further development. Among all the molecules designed, IM4 stands out as the most promising candidate. Not only does it exhibit improved anti-prostatic activity and an excellent ADME profile, but it is also the only one to show no risk of mutagenicity or carcinogenicity in predictive models. This molecule represents a particularly interesting avenue of research for future experimental validation. In summary, this theoretical study is an important step toward the design of more effective therapies for prostate cancer. Although the path to a drug is still long, this rational design methodology, combining prediction of biological activity and evaluation of ADMET properties, allows for effective screening of candidates.
Acknowledgement
The authors would like to extend their appreciation to the University Nangui ABROGOUA for the facilities it provides.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Chang, J.; Single-cell multi-stage spatial evolutional map of esophageal carcinogenesis, Cancer Cell. 2025, 43, 3, 380-397
CrossRef - Güvenç, S.; Akduran.F.; Sak. Üniversitesi Holistik Sağlık Derg. 2025,8, 61–70
CrossRef - Anusha, G.; Sunayana, R.; Ponnam, M.; Kumar, B. A, Asian J. Pharm. Res. Dev. 2020, 8, 6, 77–80
- Chhikara, B. S.; K. Parang, K. Chem. Biol. Lett. 2023, 1, 1–16
- Amaeshi, L. C.; Okunade, K. S.; Anorlu, R. I., Front. Oncol.2025, 15, 1–12
CrossRef - Zhdanovich, Y.;Ackermann, Jörg .; Wild, P.J.; Köllermann, J .; Bankov, K.; Döring, C.; Flinner, N.; Reis, H.; Wenzel, M.; Höh, B.; Mandel, P .; Vogl, T.J.; Harter, P.; Filipski, K.; Koch, I .; Bernatz, S., BMC Bioinformatics. 2023. 24, 1, 1–14
- Zafar, A.; Khatoon, S.; Khan, M. J.; Abu, J.; Naeem, A. Discov. Oncol. 2025, 16, 1
CrossRef - Adekiya, T. A., BioChem. 2025, 5, 3, 24
CrossRef - Hussein, A. M.; Badawy, N.A.; Elbayaa1, A. A.; Zaahkouk, S. A.M.; Emam, A.A.; Miski, S.F.; Belal, A.; Mehany, A.B. M., Nat. Prod. Commun. 2024, 19,12
CrossRef - Rahman, M.; Akter, K.; Kazi, R.A.; Maharub H.F.; Nahida A.; Moon, N.P.; Sang,W.S.; Bonglee, K. Cancers (Basel). 2024, 16,16, 1–36
CrossRef - Wang, R.; Chen, C .; Zhang, X .; Zhang, C.; Zhong, Q.; Chen, G .; Zhang, Qi .; Zheng, S.; Wang, G.; Chen, Q-H., J. Med. Chem.2015. 58, 11, 4713–4726
CrossRef - Do-koné, P. I. A.; N’guessan, K. N., Dembele, G. S.; Bamba, K.; Konaté, A.; Soro, D. Am. J. Chem.2025. 15, 3, 64–75
- Zak, K. M.; Waterson, A.G.; Geist, L, Braun, N, Hauer, K, Rumpel, K, Ramharter, J Stadtmueller, H .;Wolkerstorfer, B.; Schoenbauer, D.; Cui, J.; Phan, J.; Abbott, J.R.; Sarkar, D.; Hodges, T.R.; Arnold, A.; Sensintaffar, J.L.; Fesik, S.W.; Kessler, D., Am. Chem. Soc. 2024, 2
- Luttens, A .; Duy, D.; Scaletti, E.R .;Wiita, E.; Almlöf,I.; Wallner, O.; Davies.; Košenina, S.; Meng, L.; Long, M.; Mortusewicz, O.; Masuyer, G.; Ballante, F.; Michel, M.; Homan, E.; Scobie, M.; Kalderén, C.; Berglund, U.W.; Tarnovskiy, A.V .; Radchenko, D.S. .; Moroz , Y.S.; Kihlberg , J.; Stenmark, P.; Helleday,T .;Carlsson, J. Nat. Commun. 2025, l, 16, 1, 1–16
CrossRef - Dou, C. N. D.; Dembele, G.S.; Kone, M.G-R .; Tuo, N.T .; Konate, F.; Niare, A.; Karamanis, P .; Ziao, N. Comput. Chem. 2023. 11,3, 67–80
CrossRef - H. B. S .; G. E. Frisch, S. M. J. G. W. Trucks, “Gaussian 09, Revision A.02.” [Online]. Available: Gaussian, Inc., Wallingford CT, 2009.
- Ouattara, O.S .; Ziao, N., Comput. Chem. 2017, 5, 1, 38–50
CrossRef - Gramatica, P., QSAR, Comb. Sci., 2007. 26, 5, 694–701
CrossRef - Ehouman, A.; Kouakou, A.; Diarrassouba, F.; Ouattara, H. A. A.; Niamien, P. M. Orient. J. Chem. 2021, 37,805–812,
CrossRef - Shenyavskaya, E. A.; Yungman,V. S. J. Phys. Chem. Ref. Data, 2004, 33, 3, 923–957
CrossRef - Roskoski, R., Pharmacol. Res. 2023,191, 106774,
CrossRef - Nhlapho, S.; Nyathi, M.H.L.; Ngwenya , B.L.; Dube, T.; Telukdarie, A.; Munien, I.; Vermeulen, A.; Chude-Okonkwo, U.A. K.Sci. Pharm. 2024. 3,4, 177–192
CrossRef - Abdelmonem, B.H.; Abdelaal, N.M. .; Anwer, E. K. E.; Rashwan, A.A. .; Hussein, M.A.; Ahmed, Y.F.; Khashana, R.; Abdelnaser, A. , Biomedicines. 2024. 12, 7
- Fischer, B. C.; Musengi, Y.; König, J.; Sachse, B.; Hessel-Pras, S.; Schäfer, B.; Kneuer, C.; Herrmann, K., Mutagenesis. 2024, 39, 1, 32–42
CrossRef - “PreADMET | Prediction of ADME/Tox – Just another BMDRC Sites site.” Accessed: Aug. 20, 2025. [Online]. Available: https://preadmet.webservice.bmdrc.org/
Accepted on: 29 Jan 2026
Second Review by: Dr. Shreya Shanyal
Final Approval by: Dr. Murat HATİPOĞLU







ISSN Online: 2231-5039

![]()




{kind=link}