Mechanistic Insights into the Antihypertensive Effect of Resveratrol Via Inhibition of Thromboxane A2 Receptor: A Multi-Computational Approach
 and Xin Zhao1*
and Xin Zhao1*1Laboratory of Medicine, Datong Second People's Hospital (Datong Tumor Hospital), Datong, China.
2Laboratory of Molecular Imaging, Shanxi Medicine University, Taiyuan, China.
Corresponding Author E-mail: sxdteyyzx@163.com
Download this article as:
ABSTRACT:Resveratrol, a natural polyphenolic compound, was systematically investigated for its potential inhibitory interaction with the thromboxane A₂ receptor (TxA₂ R) using an integrated computational approach. ADMET prediction revealed favorable pharmacokinetic and safety profiles, including high intestinal absorption (98%), acceptable solubility, and absence of mutagenicity or hepatotoxicity, indicating good drug-likeness. DFT calculations confirmed the molecular stability and electronic properties conducive to receptor binding. Molecular docking identified a well-defined binding pose, stabilized by hydrogen bonds with THR81 and HIS89 and hydrophobic contacts with key residues. Subsequent 200 ns molecular dynamics simulations demonstrated structural stability of the complex, with consistent RMSD and compact Rg values. MM/GBSA analysis yielded a binding free energy of –26.09 kcal/mol, dominated by van der Waals and electrostatic interactions. Free energy decomposition highlighted ASP165, THR172, and GLY168 as principal contributors to binding. Hydrogen bond occupancy and free energy landscape analyses previous studies have shown that conformational stability and a persistent binding mode. Collectively, these findings indicate that resveratrol exhibits strong affinity and structural compatibility with the TxA₂ R, supporting its potential as a promising modulator for thromboxane A₂-mediated vascular dysfunction.
KEYWORDS:Free energy Calculation; Molecular docking; Molecular dynamics simulation; Resveratrol; Thromboxane A2 receptor
Introduction
Thromboxane A2 (TxA2), a metabolite of arachidonic acid, is well recognized for its ability to promote platelet aggregation and vasoconstriction through activation of TxA2/prostanoid receptor (TxA₂ R)1,2. Increasing evidence indicates that TxA₂ R plays a critical role in the pathogenesis of hypertension3. Both hypertensive patients and salt-sensitive hypertensive mouse models exhibit markedly elevated TxA2 levels accompanied by upregulated TxA₂ R expression4,5. Pharmacological blockade or genetic deletion of TxA₂ R has been shown to attenuate blood pressure elevation and vascular constriction, highlighting their pathological significance6. The three-dimensional structure of the TxA₂ R is shown in Fig S1.
Resveratrol (RSV), a natural polyphenolic compound abundant in grapes, berries, and peanuts, has attracted considerable attention due to its broad pharmacological activities, including antioxidant, anti-inflammatory, cardioprotective, and antihypertensive effects7. Accumulating evidence suggests that RSV exerts blood pressure-lowering effects through multiple mechanisms, such as improving endothelial function, enhancing nitric oxide bioavailability, and suppressing vascular oxidative stress8,9. Recent studies have further revealed that RSV can modulate arachidonic acid metabolism and downregulate the thromboxane A2 (TxA2)/TxA₂ R pathway, thereby attenuating vasoconstriction and vascular remodeling10. Given the pivotal role of TxA₂ R overactivation in the pathogenesis of hypertension5, targeting TxA₂ R signaling through RSV intervention represents a promising therapeutic strategy. Fig 1 below shows the molecular structure of RSV.
This study applied a comprehensive computational approach to predict the inhibitory interaction between resveratrol and the thromboxane A₂ receptor (TxA₂ R). The aim was to establish a theoretical framework for identifying natural compounds with potential antihypertensive activity through receptor modulation. Compared with conventional drug discovery strategies, computational simulations provide clear advantages in characterizing binding mechanisms, optimizing molecular conformations, and accelerating the identification of promising candidates. By reducing the number of compounds subjected to subsequent laboratory evaluation, in silico screening enhances both the efficiency and cost-effectiveness of early-stage drug discovery.
 |
Figure 1: Molecular structure of resveratrol Click here to View Figure |
Materials and Methods
Prediction of ADMET Profile
Inefficient pharmacokinetics and unexpected toxicity are among the predominant causes of attrition in the development of new therapeutic agents. Implementing computational prediction at the preliminary stage provides an effective means of improving the likelihood of success in drug discovery pipelines. In this context, before proceeding with molecular docking and structural refinement, we systematically evaluated the pharmacokinetic characteristics, safety profile, and overall drug-likeness of resveratrol by employing three widely accessible in silico platforms: pk CSM11 (https://biosig.lab.uq.edu.au/pkcsm/), Tox ACoL12 (https://toxacol.bioinforai.tech/), and the Swiss ADME database13 (http://www.swissadme.ch). Critical descriptors, including absorption, distribution, metabolism, excretion, compliance with Lipinski’s Rule of Five, and predicted toxicity, were analyzed, thereby providing essential insights into whether resveratrol demonstrates physicochemical properties aligned with clinically validated drugs.
pKa calculation
The prediction of pKa values offers significant advantages, as it enables the rapid and accurate determination of the ionization state of compounds, which is crucial in drug development research14. In this study, the pKa values of resveratrol were calculated using Chemaxon software based on the distribution of partial charges within the molecule. Furthermore, the relative proportions of different ionization states were determined under various pH conditions, providing insights into the compound’s physicochemical behavior in biological environments.
Conformational search
Computational conformer generation and energy minimization allow rapid and accurate exploration of a large number of possible molecular structures, enabling identification of low-energy conformations in a short time15. In this study, 50 distinct conformers of resveratrol were generated using RDKit16,17, and the lowest-energy structure was selected for subsequent investigation of its potential interaction with the thromboxane A2 receptor.
Density functional theory calculations
To investigate the structural and electronic features of resveratrol and their potential role in modulating the thromboxane A₂ receptor (TxA₂ R), a systematic computational study was conducted using the density functional theory (DFT) approach. The molecular geometry of resveratrol was first optimized at the B3LYP/6-311++G** level to obtain the most stable low-energy conformation. Subsequent frequency analysis confirmed the absence of imaginary frequencies, verifying that the optimized structure represents a true local minimum with thermodynamic stability. Based on this optimized geometry, frontier molecular orbital (FMO) characteristics and molecular electrostatic type (MET) charge distribution were calculated to elucidate the electronic properties and potential binding reactivity of resveratrol.
Molecular docking
To construct the initial model of the resveratrol–receptor complex, molecular docking was performed using Auto Dock 4.2. The ligand structure was obtained from the lowest-energy conformation generated by density functional theory (DFT) optimization, while the TxA₂ R structure was retrieved from the Protein Data Bank (PDB, ID:6llU)18 with a defined resolution, representing the crystallographic structure of the thromboxane A₂ receptor. Receptor and ligand preparation were carried out using Auto Dock Tools 1.5.6. In the preprocessing procedure, all co-crystallized ligands and water molecules were removed, and polar hydrogens together with Gasteiger charges were added to the receptor to improve docking accuracy. A three-dimensional docking grid of 40 × 40 × 40 points with a spacing of 0.375 Å was applied to encompass the putative binding pocket. The docking calculations employed the Lamarckian genetic algorithm (LGA) implemented19 in Auto Dock 4.2, generating 2000 potential binding poses. The most favorable conformation was chosen based on its binding orientation within the active site as well as the corresponding binding energy, which was subsequently used as the starting point for further structural and mechanistic analysis.
MD simulations
In the molecular dynamics (MD) simulations, the most favorable resveratrol-TxA₂ R binding pose obtained from docking was employed as the initial structural model. Ligand parameters were generated according to the General AMBER Force Field (GAFF 2.0)20, with Antechamber in Amber Tools used to derive atomic charges and topology files. The TxA₂ R and its binding interface with resveratrol were described using the Amber ff19SB force field21. Each complex system was subjected to 200 ns of all-atom MD simulations using the GPU-accelerated PMEMD.CUDA engine implemented in AMBER 2222, ensuring adequate conformational sampling. Post-simulation analyses were performed with CPPTRAJ23,24, including assessments of system energy convergence, backbone and side-chain flexibility, as well as dynamic interaction patterns of residues within the active binding region.
Binding free energy calculation
To quantitatively evaluate the binding strength between resveratrol and the thromboxane A₂ receptor (TxA₂ R), the binding free energy was estimated using the molecular mechanics/generalized Born surface area (MM/GBSA) approach25. This method is commonly employed due to its balance of computational efficiency and reliability in predicting receptor-ligand interaction energies. The calculation was performed on trajectory snapshots extracted from the last 20 ns of the molecular dynamics simulations, with energy values averaged over 2000 representative frames. In addition, per-residue free energy decomposition was conducted using the MM/GBSA protocol to reveal the energetic contributions of specific amino acid residues to ligand binding. All computations were executed using the MMPBSA.py module in the AMBER software suite26.
Free energy landscape analysis
To explore the conformational states of resveratrol during molecular dynamics simulations, the trajectory files were first preprocessed using the CPPTRAJ module implemented in AMBER22. The cleaned trajectories were then analyzed with GROMACS utilities to calculate the RMSD and radius of Rg over a 200 ns simulation. These parameters were employed as reaction coordinates for constructing the free energy landscape (FEL). The resulting data were imported into Origin 2021 for visualization, where three-dimensional FEL plots were generated to represent the energy distribution of receptor conformations along the RMSD and Rg axes27.
Results
Application of computational ADMET data prediction
In the context of drug discovery, computational approaches for predicting ADMET properties are indispensable for early-stage evaluation of drug candidates28. Lipinski’s “Rule of Five” remains a widely applied benchmark for assessing oral drug-likeness29. The calculation results of this time are shown in Table 1. Resveratrol meets all the standards of Lipinski, indicating that it has good potential for oral bioavailability. Among the pharmacokinetic parameters, water solubility is a critical determinant of absorption and systemic distribution. Resveratrol shows a predicted Log S of –3.36, placing it within the moderately soluble range (–4 to –2), which is considered acceptable for oral formulations. It’s predicted human intestinal absorption rate reaches 98%, reflecting efficient uptake through the gastrointestinal tract and further supporting its oral absorption potential.
Regarding distribution, the steady-state volume of distribution (VDss) exhibits a Log value of –0.15, implying that resveratrol predominantly resides within the plasma compartment with limited tissue penetration. Nonetheless, its blood–brain barrier (BBB) permeability index of 0.97 suggests a pronounced ability to cross into the central nervous system, highlighting possible therapeutic applications in neurological or cerebrovascular disorders.
The metabolism-related predictions show that resveratrol has a high likelihood of interaction with CYP450 1A2 (0.97), while exhibiting a moderate probability of being a substrate for CYP450 2D6 (0.46). This implies that phase I metabolic transformation may be primarily mediated through specific CYP isoforms. In terms of excretion, the compound demonstrates a total clearance rate of 0.91 ml/min/kg, indicating efficient systemic elimination, while its renal OCT2 substrate probability remains relatively low (0.10), suggesting that non-renal pathways play a larger role in clearance.
Toxicity predictions further support a favorable safety profile: resveratrol is considered negative for AMES mutagenicity, non-sensitizing to skin, and non-hepatotoxic. Additionally, the maximum tolerated dose (MTD) is predicted to be 1.2 log mg/kg/day, reflecting good oral tolerance and low toxicity risk.
Collectively, these ADMET results underline that resveratrol possesses promising pharmacokinetic and toxicological characteristics, with suitable solubility, high gastrointestinal absorption, minimal predicted toxicity, and substantial BBB permeability. These properties make resveratrol a compelling candidate for oral drug development and justify its further exploration in preclinical and clinical studies.
Table 1: Data on pharmacokinetics and toxicity properties obtained from Swiss ADME, ADMET SAR, and pK CSM online servers.
| Molecules | Resveratrol | |
| AMES Toxicity | Safe | |
| Absorption | Water Solubility Log S | -3.36 |
| Human Intestinal Absorption (%) | 0.98 | |
| Distribution | VDss (Log L/kg) | -0.15 |
| BBB Permeability | 0.97 | |
| Metabolism | CYP450 1A2 Inhibitor | 0.97
(Inhibitor) |
| CYP450 2D6 Substrate | 0.46
(Non-substrate) |
|
| Excretion | Total Clearance (ml/min/kg) | 0.91 |
| Renal OCT2 Substrate | 0.10 | |
| Toxicity | Max. Tolerated Dose (log mg/kg/day) | 1.2 |
| Skin Sensitization | Safe | |
| Hepatotoxicity | Safe | |
| Lipinski Rule | Result | Yes |
| Violation | 0 | |
pKa calculation
To better characterize the ionization behavior of resveratrol, the pKa values of its three phenolic hydroxyl groups were theoretically estimated. As shown in Fig S2, the calculated pKa values were 8.3, 8.9, and 9.9, respectively. As shown in Fig S3, these results suggest that resveratrol predominantly exists in its neutral form (approximately 90%) under physiological pH, which may facilitate favorable interactions with the thromboxane A₂ receptor.
Conformational search
To explore the structural flexibility of resveratrol, a conformational search was performed using RDKit30, which generated 50 distinct conformers (Fig S4). After subsequent geometry optimization, their relative energies were evaluated. The energy distribution indicated that most conformers were clustered within a narrow energy window of less than -765 hartree, reflecting limited conformational diversity. Among these, the global minimum conformer was identified as the most energetically favorable structure and was therefore selected for subsequent molecular docking and dynamic simulations. This strategy ensured that the computational analyses were based on the most stable and representative conformation, thereby improving the reliability of the predicted binding interactions with the thromboxane A₂ receptor.
DFT calculation studies
To better understand the structural and electronic properties of resveratrol, density functional theory (DFT) calculations were carried out at the B3LYP/6-311++G** level of theory31. The optimized geometry of the equilibrium structure is shown in Fig S5. The thermodynamic descriptors, dipole moment, and polarizability obtained from these calculations are summarized in Table 2.
The calculated dipole moment (µ) of resveratrol is 2.76 Debye, which indicates a moderate degree of molecular polarity and suggests the potential to form hydrogen bonds with polar residues within the thromboxane A₂ receptor binding pocket. The static polarizability (α) is 184.55 Bohr³, reflecting a significant electron cloud flexibility that can enhance intermolecular interactions through dispersion forces and facilitate adaptive binding to diverse receptor microenvironments.
Thermodynamic energy parameters were also derived, including the zero-point vibrational energy (ZPVE, –766.13 Hartree), the total electronic and thermal energy (Etot, –766.12 Hartree), the enthalpy (H, –766.12 Hartree), and the Gibbs free energy (G, –766.18 Hartree). These values highlight the thermodynamic stability of resveratrol in its optimized state, supporting its suitability as a drug-like molecule.
Table 2: Thermal parameters (Hartree/particle), polarizabilities (Bohr3), and dipole moments (Debye)of resveratrol.
| Parameter | Resveratrol |
| Ecorr | 0.23 |
| ZPVE | -766.13 |
| Etot | -766.12 |
| H | -766.12 |
| G | -766.18 |
| Total Dipole Moment µ | 2.76 |
| Polarizability α | 184.55 |
Ecorr: zero-point correction; ZPVE: sum of electronic and zero-point energies; Etot: sum of electronic and thermal energies; H: sum of electronic and thermal enthalpies; G: sum of electronic and thermal free energies.
Molecular Electrostatic Potential (MEP)
The molecular electrostatic potential (MEP) surface of resveratrol, displayed in Fig S6, provides insights into its reactive regions. Red regions correspond to high electron density, indicating preferred sites for electrophilic attack, whereas blue areas reflect electron-deficient zones favorable for nucleophilic interaction. The MEP profile shows pronounced negative potential around the hydroxyl oxygen atoms, consistent with their ability to participate in hydrogen bonding, which may contribute to receptor binding affinity.
Frontier Molecular Orbitals (FMOs)
The frontier molecular orbital (FMO) analysis of resveratrol revealed that the HOMO energy level is –7.49 eV, while the LUMO lies at 2.52 eV, resulting in an energy gap (ΔE) of approximately 10.01 eV (as shown in Fig S7). The relatively large HOMO–LUMO separation suggests that resveratrol possesses high chemical stability and moderate reactivity. Spatial distribution analysis showed that the HOMO density is primarily localized on the aromatic rings and hydroxyl groups, indicating strong electron-donating potential, whereas the LUMO is delocalized over the π-conjugated system, reflecting its electron-accepting ability. This electronic structure highlights resveratrol’s potential to participate in noncovalent interactions within the thromboxane A₂ receptor binding site, which may contribute to its inhibitory activity.
Molecular Docking Study
Molecular docking was conducted to characterize the interaction between resveratrol and the target receptor. All generated docking conformations were clustered into a single group, indicating a highly consistent binding orientation. The docking scores of these poses were all below -5.0 kcal/mol, reflecting a strong binding tendency of resveratrol to the protein.
As shown in Fig 2A, resveratrol was positioned within the binding pocket, where it established stable hydrogen bonds with residues THR81 and HIS89. In addition, the 2D Lig Plot representation (Fig 2B) revealed hydrophobic contacts with several surrounding residues, including SER181, VAL85, TRP182, and MET112, further supporting the stability of the complex. These results demonstrate that resveratrol occupies the catalytic cavity and engages in both hydrogen bonding and hydrophobic interactions with key residues. The uniformity of docking conformations further suggests that resveratrol has a well-defined binding mode. Nonetheless, since docking describes a static interaction, molecular dynamics simulations are required to validate the persistence of these interactions over time.
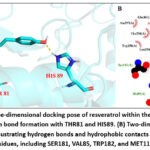 |
Figure 2: (A) Three-dimensional docking pose of resveratrol within the binding pocket, showing hydrogen bond formation with THR81 and HIS89. (B) Two-dimensional Lig Plot representations illustrating hydrogen bonds and hydrophobic contacts with surrounding residues, including SER181, VAL85, TRP182, and MET112. |
MD Simulation
To evaluate the stability of the resveratrol–thromboxane A₂ receptor complex, a 200 ns molecular dynamics (MD) simulation was carried out using the AMBER suite. As this timescale is commonly sufficient for protein–ligand complexes to reach structural equilibration and produce statistically reliable binding stability parameters, according to previous studies. As shown in Fig 3A, the backbone RMSD of the receptor gradually reached equilibrium after the initial fluctuations and remained relatively stable, indicating that the overall fold of the protein was preserved. In contrast, resveratrol displayed lower RMSD values throughout the trajectory, suggesting that the ligand maintained a consistent binding orientation within the active site.
Further insights were obtained from the RMSF profile of receptor residues (Fig 3B). Most residues exhibited only minor fluctuations (<2.5 Å), while a few loop regions displayed higher flexibility, consistent with their dynamic nature. Notably, the transmembrane helices remained structurally stable, demonstrating that ligand binding did not induce large conformational changes in the receptor. These results collectively support that resveratrol can form a stable complex with the thromboxane A₂ receptor, with structural dynamics largely confined to peripheral flexible segments rather than the ligand-binding core.
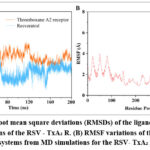 |
Figure 3: (A) Root mean square deviations (RMSDs) of the ligand during 200 ns MD simulations of the RSV – TxA₂ R. (B) RMSF variations of the Cα atom of complexed systems from MD simulations for the RSV- TxA₂ R systems. |
To further characterize the dynamic behavior of the resveratrol–thromboxane A₂ receptor complex, additional structural parameters were analyzed. The Rg remained stable across the 200 ns trajectory, suggesting that the global compactness of the receptor was preserved upon ligand binding(Fig S8). Similarly, the SASA exhibited only minor fluctuations, and the ligand consistently showed a low SASA value, indicating that resveratrol was stably buried within the binding pocket without undergoing significant solvent exposure(Fig S9).
Finally, representative conformations were extracted at 50, 100, 150, and 200 ns. As illustrated in Fig S10, the overall architecture of the receptor remained essentially unchanged throughout the simulation, and the binding orientation of resveratrol within the active site was consistently maintained. No significant structural rearrangements of the receptor or substantial displacement of the ligand were observed, underscoring the robustness and consistency of the binding mode.
Binding free energy
The binding free energy serves as a quantitative indicator of the strength of interaction between resveratrol and the thromboxane A₂ receptor (TxA₂ R). As summarized in Table S2, the calculated binding free energy for the resveratrol – TxA₂ R complex was −26.09 kcal/mol, suggesting a stable and energetically favorable interaction. Decomposition of the energy terms revealed that van der Waals forces (EvdW = −29.50 kcal/mol) make a substantial contribution to the binding, highlighting the importance of hydrophobic contacts in stabilizing the complex. Electrostatic interactions (Eele = −19.88 kcal/mol), although weaker compared to van der Waals forces, also provided favorable contributions. The polar solvation term (EGB = 27.80 kcal/mol) exerted a destabilizing effect, which partially counteracted the favorable electrostatic interactions, a phenomenon commonly observed in ligand–protein complexes. In contrast, the non-polar solvation energy (Esurf = −4.51 kcal/mol) contributed slightly to the stabilization. Overall, the negative free energy value indicates that resveratrol binds to A₂R with high affinity, with both hydrophobic and electrostatic forces working cooperatively to stabilize the ligand within the binding pocket. This result suggests that resveratrol has the potential to act as a strong inhibitor by maintaining a stable interaction with the thromboxane A₂ receptor.
Free energy decomposition
To identify the principal residue determinants that stabilize the resveratrol–thromboxane A₂ receptor complex, per-residue MM/GBSA decomposition was performed. The decomposition outcomes (Fig 4 and Table) indicate that individual residues make modest but meaningful contributions to binding, with per-residue subtotal free energies ranging from approximately −1.6 to −1.0 kcal·mol⁻¹ for the most influential sites. Notably, ASP165, THR172, and GLY168 each contribute ~−1.5 to −1.6 kcal·mol⁻¹ to the interaction, highlighting a cluster of residues near the binding pocket that collectively favor ligand association. A closer inspection of the energy components reveals distinct physical origins for these contributions. Several residues display negative van der Waals terms (∆EvdW ≈ −0.9 to −1.3 kcal·mol⁻¹), indicating that hydrophobic contacts and close packing are important stabilizing forces. In contrast, strongly favorable electrostatic terms (for example, the large negative ∆Eele for ASP165) are frequently offset by unfavorable polar solvation (∆Gsol,GB positive), resulting in only a small net polar contribution at the residue level. This compensation between direct Coulombic interactions and desolvation penalties is a typical feature of protein–ligand complexes in aqueous environments. Overall, the decomposition points to a binding mode dominated by nonpolar interactions at the residue level, supplemented by localized electrostatic effects that are partially neutralized by solvent screening. From an optimization standpoint, these findings suggest that enhancing hydrophobic complementarity within the identified pocket is a promising strategy to increase binding affinity, while modifications aimed solely at strengthening polar contacts must account for the associated solvation cost.
 |
Figure 4: (A) The binding free energy of RSV – TxA₂ R (B) The free energy of RSV – TxA₂ R dissociation Click here to View Figure |
Analysis of hydrogen bond and free energy landscapes
To further elucidate the stability and interaction pattern of the resveratrol–TxA₂ R complex, hydrogen bond dynamics and free energy landscapes (FEL) were analyzed over the course of the 200 ns molecular dynamics simulation (Fig 5 and Fig S11). Hydrogen bonds were monitored between resveratrol and individual receptor residues, with occupancy values representing the proportion of simulation time during which the bonds were maintained (Table S1). The results indicated that resveratrol consistently formed hydrogen bonds with MET203 (30.62%) and SER272 (29.77%), suggesting that these two residues play a pivotal role in maintaining the ligand–receptor complex. The presence of multiple hydrogen bonding events throughout the trajectory highlights the dynamic yet stable nature of these interactions. Moreover, the number of hydrogen bonds fluctuated primarily between 1 and 3 during the simulation, which is characteristic of a moderately strong and persistent binding mode (Fig. S1). To further explore the conformational energy profile, a Gibbs free energy landscape (FEL) was constructed using RMSD and Rg as reaction coordinates (Fig. 5A and Fig. 5B). The FEL revealed a dominant global minimum region located at approximately RMSD = 2.3 Å and Rg = 23.5 Å, corresponding to the most stable conformational ensemble of the resveratrol–TxA₂ R complex. The narrow and deep energy basin indicated limited structural fluctuations and high conformational stability of the complex during the equilibrium phase. The continuous and smooth energy surface, previous studies have shown that resveratrol binding does not induce significant conformational rearrangements in the receptor structure, but rather stabilizes its active conformation. Collectively, the hydrogen bond occupancy and FEL analyses suggest that resveratrol forms a stable complex with the TxA₂ R, mainly stabilized by persistent hydrogen bonds and compact tertiary structure. The consistent energy minimum observed throughout the simulation supports the notion that resveratrol maintains strong structural complementarity with the receptor binding pocket, reinforcing its potential as a viable TxA₂ R modulator.
 |
Figure 5: (A) Two-dimensional projection of the principal eigenvectors (RMSD and Rg) from the initial position to 200 nanoseconds of molecular dynamics simulation. (B) 3D free energy landscape diagram. |
Conclusions
Hypertension, a chronic cardiovascular disorder characterized by vascular remodeling and endothelial dysfunction32, continues to be a major global health concern33. Accumulating evidence indicates that activation of the thromboxane A₂ receptor (TxA₂ R) is critically involved in vasoconstriction and platelet aggregation, thereby contributing to elevated vascular resistance and inflammation. Hence, targeting the TxA₂ R has emerged as a promising therapeutic strategy for hypertension management34-36. In this study, an in silico approach combining ADMET prediction, quantum chemical analysis, molecular docking, and long-timescale MD simulations was employed to elucidate the interaction mechanism and dynamic stability of resveratrol with the TxA₂ R.
The ADMET assessment revealed that resveratrol possesses favorable pharmacokinetic properties, including high intestinal absorption, low hepatotoxicity, and acceptable compliance with Lipinski’s rule of five. The compound exhibited moderate hydrophilicity (Log S = -3.36) and strong oral bioavailability, suggesting that it could achieve adequate systemic exposure in vivo. Toxicity predictions using the Tox ACoL platforms indicated that resveratrol belongs to a safe toxicity class, with a high LD₅₀ threshold, confirming its low probability of acute or chronic toxicity. These findings provide initial pharmacological support for the further development of resveratrol as a TxA₂ R antagonist.
The molecular geometry of resveratrol was optimized using density functional theory (DFT) at the B3LYP/6-311++G** level, yielding a stable low-energy structure confirmed by the absence of imaginary frequencies. The frontier molecular orbital (FMO) analysis showed a HOMO–LUMO energy gap of 10.01 eV, indicating moderate molecular reactivity and electronic stability. The molecular electrostatic potential (MEP) map suggested that the phenolic hydroxyl groups represent the major electron-donating regions, which may facilitate hydrogen bonding with polar residues within the receptor active site.
To explore the stability of the resveratrol-TxA₂ R complex at the atomic level, a 200-nanosecond molecular dynamics simulation was conducted using the AMBER 22 software suite. The root mean square deviation (RMSD) curve indicated that the complex remained stable throughout the simulation. The root mean square fluctuation (RMSF) analysis revealed a reduction in the flexibility of the residues within the binding region, suggesting that ligand binding restricted local conformational movements. The radius of gyration (Rg) also remained stable, while the solvent-accessible surface area (SASA) slightly decreased, indicating that the complex configuration became more compact and energetically stable after resveratrol binding. Binding free energy calculations using the MM/GBSA approach, previous studies have shown that the thermodynamic stability of the complex has been established. The total binding energy (ΔGbind) of resveratrol was −26.09 kcal/mol, dominated by van der Waals (−29.05 kJ/mol) and electrostatic (−19.88 kJ/mol) contributions, while the polar solvation term opposed complex formation. Per-residue decomposition analysis identified ASP 165, THR 172, and GLY 168 as key contributors to binding affinity, consistent with the docking interaction map. This agreement between static docking and dynamic free energy evaluation supports the reliability of the binding mechanism. The free energy landscape (FEL) analysis, constructed using RMSD and Rg as reaction coordinates, revealed a well-defined, single global minimum basin for the resveratrol–TxA₂ R complex, indicative of a highly stable conformational state. In contrast, the apo receptor displayed a broader, multi-basin energy surface, reflecting enhanced flexibility and lower conformational stability. This suggests that resveratrol binding effectively restricts large-scale conformational fluctuations and stabilizes the receptor in a compact, energetically favorable configuration.
Collectively, the results demonstrate that resveratrol exhibits excellent pharmacokinetic behavior, favorable electronic characteristics, and robust binding affinity toward the TxA₂ R. The compound not only engages in strong hydrogen bonding and hydrophobic contacts with critical residues but also maintains structural integrity and thermodynamic stability throughout extended molecular simulations. These in silico findings provide compelling evidence that resveratrol can function as a potent natural antagonist of the TxA₂ R, thereby offering mechanistic insight into its reported antihypertensive and vasoprotective effects. Further in vitro and in vivo investigations are warranted to confirm these computational predictions and explore their therapeutic potential in cardiovascular disease intervention.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Sstatement
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
Ethics Statement
This study does not require ethical approval as it only involves computer simulation methods, including molecular docking, molecular dynamics simulation, and free energy calculation, and does not involve human or animal subjects.
Informed Consent Statement
As this study is based on computational methods and does not involve human or animal data, obtaining informed consent was not applicable
Author Contributions
ZX contributed to conceptualization, investigation, methodology, data organization, visualization, formal analysis, and drafting of the original manuscript. ZX also participated in manuscript review and editing, funding acquisition, and project supervision. Y Z made substantial contributions, including extensive manuscript polishing, language editing, and refinement of scientific expression, which significantly improved the clarity and overall quality of the article.
References
- Cheng, Y. et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science (New York, N.Y.) 296, 539-541, doi:10.1126/science.1068711 (2002).
CrossRef - Tomaiuolo, M., Brass, L. F. & Stalker, T. J. Regulation of Platelet Activation and Coagulation and Its Role in Vascular Injury and Arterial Thrombosis. Interventional cardiology clinics 6, 1-12, doi:10.1016/j.iccl.2016.08.001 (2017).
CrossRef - Eckenstaler, R. & Benndorf, R. A. Insights into the Expression, Structure, and Function of the Thromboxane A2 Receptor in Vascular Biology. Acs Pharmacology & Translational Science 8, 2887-2907, doi:10.1021/acsptsci.5c00378 (2025).
CrossRef - Cavka, A. et al. The role of cyclo-oxygenase-1 in high-salt diet-induced microvascular dysfunction in humans. The Journal of physiology 593, 5313-5324, doi:10.1113/jp271631 (2015).
CrossRef - Capra, V., Bäck, M., Angiolillo, D. J., Cattaneo, M. & Sakariassen, K. S. Impact of vascular thromboxane prostanoid receptor activation on hemostasis, thrombosis, oxidative stress, and inflammation. Journal of thrombosis and haemostasis : JTH 12, 126-137, doi:10.1111/jth.12472 (2014).
CrossRef - Braun, H. et al. Deletion of vascular thromboxane A(2) receptors and its impact on angiotensin II-induced hypertension and atherosclerotic lesion formation in the aorta of Ldlr-deficient mice. Biochemical pharmacology 219, 115916, doi:10.1016/j.bcp.2023.115916 (2024).
CrossRef - Yu, X., Jia, Y. & Ren, F. Multidimensional biological activities of resveratrol and its prospects and challenges in the health field. Front Nutr 11, 1408651, doi:10.3389/fnut.2024.1408651 (2024).
CrossRef - Parsamanesh, N. et al. Resveratrol and endothelial function: A literature review. Pharmacological research 170, 105725, doi:10.1016/j.phrs.2021.105725 (2021).
CrossRef - Wang, L. P. et al. Resveratrol reduces RVLM neuron activity via activating the AMPK/Sirt3 pathway in stress-induced hypertension. The Journal of biological chemistry 301, 108394, doi:10.1016/j.jbc.2025.108394 (2025).
CrossRef - Li, Q. et al. Resveratrol attenuates cyclosporin A-induced upregulation of the thromboxane A(2) receptor and hypertension via the AMPK/SIRT1 and MAPK/NF-κB pathways in the rat mesenteric artery. Eur J Pharmacol 972, 176543, doi:10.1016/j.ejphar.2024.176543 (2024).
CrossRef - Pires, D. E., Blundell, T. L. & Ascher, D. B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. Journal of medicinal chemistry 58, 4066-4072, doi:10.1021/acs.jmedchem.5b00104 (2015).
CrossRef - Lu, J. et al. ToxACoL: an endpoint-aware and task-focused compound representation learning paradigm for acute toxicity assessment. Nature communications 16, 5992, doi:10.1038/s41467-025-60989-7 (2025).
CrossRef - Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific reports 7, 42717, doi:10.1038/srep42717 (2017).
CrossRef - Yang, C. et al. In silico prediction of pK (a) values using explainable deep learning methods. Journal of pharmaceutical analysis 15, 101174, doi:10.1016/j.jpha.2024.101174 (2025).
CrossRef - McNutt, A. T. et al. Conformer Generation for Structure-Based Drug Design: How Many and How Good? J Chem Inf Model 63, 6598-6607, doi:10.1021/acs.jcim.3c01245 (2023).
CrossRef - Tosco, P., Stiefl, N. & Landrum, G. Bringing the MMFF force field to the RDKit: implementation and validation. Journal of cheminformatics 6, doi:10.1186/s13321-014-0037-3 (2014).
CrossRef - Bento, A. P. et al. An open source chemical structure curation pipeline using RDKit. Journal of cheminformatics 12, doi:10.1186/s13321-020-00456-1 (2020).
CrossRef - Fan, H. et al. Structural basis for ligand recognition of the human thromboxane A(2) receptor. Nature chemical biology 15, 27-33, doi:10.1038/s41589-018-0170-9 (2019).
CrossRef - Bitencourt-Ferreira, G., Pintro, V. O. & de Azevedo, W. F., Jr. Docking with AutoDock4. Methods in molecular biology (Clifton, N.J.) 2053, 125-148, doi:10.1007/978-1-4939-9752-7_9 (2019).
CrossRef - Shivakumar, D. et al. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. Journal of chemical theory and computation 6, 1509-1519, doi:10.1021/ct900587b (2010).
CrossRef - Tian, C. et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. Journal of chemical theory and computation 16, 528-552, doi:10.1021/acs.jctc.9b00591 (2020).
CrossRef - Peramo, A. Solvated and generalised Born calculations differences using GPU CUDA and multi-CPU simulations of an antifreeze protein with AMBER. Molecular Simulation 42, 1263-1273, doi:10.1080/08927022.2016.1183000 (2016).
CrossRef - Roe, D. R. & Cheatham, T. E. Parallelization of CPPTRAJ Enables Large Scale Analysis of Molecular Dynamics Trajectory Data. Journal of computational chemistry 39, 2110-2117, doi:10.1002/jcc.25382 (2018).
CrossRef - Roe, D. R. & Cheatham, T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. Journal of chemical theory and computation 9, 3084-3095, doi:10.1021/ct400341p (2013).
CrossRef - Wang, E. C. et al. VAD-MM/GBSA: A Variable Atomic Dielectric MM/GBSA Model for Improved Accuracy in Protein-Ligand Binding Free Energy Calculations. Journal of Chemical Information and Modeling 61, 2844-2856, doi:10.1021/acs.jcim.1c00091 (2021).
CrossRef - Miller, B. R. et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of chemical theory and computation 8, 3314-3321, doi:10.1021/ct300418h (2012).
CrossRef - Bai, H. et al. Machine learning-based QSAR and structure-based virtual screening guided discovery of novel mIDH1 inhibitors from natural products. J Comput Aided Mol Des 39, 44, doi:10.1007/s10822-025-00624-1 (2025).
CrossRef - Azad, I. et al. Identification of Severe Acute Respiratory Syndrome Coronavirus-2 inhibitors through in silico structure-based virtual screening and molecular interaction studies. Journal of molecular recognition : JMR 34, e2918, doi:10.1002/jmr.2918 (2021).
CrossRef - Permana, A. et al. Virtual Screening, Toxicity Evaluation and Pharmacokinetics of Erythrina Alkaloids as Acetylcholinesterase Inhibitor Candidates from Natural Products. Advances and applications in bioinformatics and chemistry : AABC 17, 179-201, doi:10.2147/aabc.S495947 (2024).
CrossRef - Spiekermann, K., Pattanaik, L. & Green, W. H. High accuracy barrier heights, enthalpies, and rate coefficients for chemical reactions. Scientific data 9, 417, doi:10.1038/s41597-022-01529-6 (2022).
CrossRef - Sripadung, P., Rajchakom, C., Nunthaboot, N., Jiang, X. & Sungthong, B. Computational and Experimental Insights into Tyrosinase and Antioxidant Activities of Resveratrol and Its Derivatives: Molecular Docking, Molecular Dynamics Simulation, DFT Calculation, and In Vitro Evaluation. International journal of molecular sciences 26, doi:10.3390/ijms26188827 (2025).
CrossRef - Gallo, G., Volpe, M. & Savoia, C. Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front Med (Lausanne) 8, 798958, doi:10.3389/fmed.2021.798958 (2021).
CrossRef - Humphrey, J. D. Mechanisms of Vascular Remodeling in Hypertension. American journal of hypertension 34, 432-441, doi:10.1093/ajh/hpaa195 (2021).
CrossRef - Davì, G., Santilli, F. & Vazzana, N. Thromboxane receptors antagonists and/or synthase inhibitors. Handbook of experimental pharmacology, 261-286, doi:10.1007/978-3-642-29423-5_11 (2012).
CrossRef - Ramos-Alves, F. E., de Queiroz, D. B., Santos-Rocha, J., Duarte, G. P. & Xavier, F. E. Increased cyclooxygenase-2-derived prostanoids contributes to the hyperreactivity to noradrenaline in mesenteric resistance arteries from offspring of diabetic rats. PLoS One 7, e50593, doi:10.1371/journal.pone.0050593 (2012).
CrossRef - Allen, M. F. et al. Difluorinated thromboxane A(2) reveals crosstalk between platelet activatory and inhibitory pathways by targeting both the TP and IP receptors. British journal of pharmacology 181, 3685-3699, doi:10.1111/bph.16435 (2024).
CrossRef
Second Review by: Dr. Rashim Mohhamad
Final Approval by: Dr. Tanay Pramanik








![]()




{kind=link}