Rational Design and Computational Evaluation Of Sulfonamide-Quinoline Derivatives as Multi-Targeted Anti-Cancer Agents
 , Kajal Kaushik and Tripti Arora*
, Kajal Kaushik and Tripti Arora*SGT College of Pharmacy, SGT University, Gurugram, Haryana, India.
Corresponding Author E-mail:aroratripti.6.9@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/410506
Download this article as:
![]()
Cancer is a principal cause of mortality globally, driven by genetic and epigenetic modifications that result in unregulated cellular proliferation. As a result of the shortcomings of current treatments, such as drug resistance and toxicity, this study focuses on the rational design of four A, B, C and D series of novel sulfonamide-quinoline derivatives bearing different amino linkers targeting key cancer-associated enzymes: carbonic Anhydrase IX (CAIX), Aurora Kinase A (AURKA), and Aurora Kinase B (AURKB). Molecular docking studies revealed B5, B3, C5, C3, A5, and D2 as the most promising inhibitors that showed strong binding affinities for all three target enzymes. In addition, A2, A6, B6, D3, and D6 showed selective inhibition of AURKA and AURKB. Calculations of the pharmacokinetic parameters of the hybrids were also performed, which showed favorable results. Toxicity profiling showed that A2, B4, B5, B6, D4, D5, and D6 were less toxic than Sitamaquine, but all needed optimization except for respiratory toxicity. Most compounds had good GI absorption, while B4, D4, D5, and D6 would likely require formulation adjustment to enhance their bioavailability. This study indicates that sulfonamide-quinoline derivatives are promising multi-target anti-cancer agents and deserve further in vitro and in vivo studies.
KEYWORDS:Aurora Kinase A; Aurora Kinase B; Carbonic Anhydrase IX, Docking; Quinoline; Sulfonamide
Introduction
Cancer is a genetic disease that can spread throughout the body and is characterised by aberrant cell proliferation and genetic or epigenetic alterations in somatic cells.1 Globally, there were 9.6 million cancer-related deaths and 18 million cancer diagnoses in 2018. It is anticipated that there would be 420 million cases annually by 2025.2,3 Prostate, colorectal, lung, and breast cancers are among the common varieties.[4] Drug resistance and serious adverse effects (such as nausea, hair loss, and immunological suppression) are problems with current treatments. These problems show how urgently safer, more potent, and less toxic anti-cancer medications are needed.5 In medicinal chemistry, molecular hybridization—the logical fusion of two or more pharmacophoric moieties into a single hybrid structure—has become a potent tactic. Compounds with increased biological activity, better selectivity, and the ability to overcome drug resistance are frequently the outcome of this strategy.6
For many years, quinoline has been an essential scaffold in drug development, particularly in anticancer studies. Quinoline is a nitrogen-based heterocyclic moiety with biological action. Nitrogen atoms greatly enhance the basic nature of quinoline compounds. Several anticancer medicines based on the quinoline framework are in clinical testing. Quinoline derivatives inhibit several cancer-causing enzymes, such as tyrosine kinase, PI3K-pKB (phosphoinositide 3-kinase-protein kinase B), epidermal growth factor receptors, ALK5(Activin-receptor-like kinase-5), mitogen-activated protein kinase, platelet-d kinase insert domain receptors, and nonreceptor tyrosine kinases. Quinoline-based anticancer medicines, including Bosutinib, Lenvatinib, and Cabozantinib, prevent protein kinase activity.[7]
Quinoline derivatives have shown promising activity in many cancer cell lines, including colorectal, breast, colon, lung, and renal. [8-20]
In medicinal chemistry, the sulfonamide moiety has gained popularity, leading to the creation of various sulfonamide derivatives with various biological actions. These actions include anti-oxidant,[22] anti-bacterial,[23] anti-fungal,[24] anti-inflammatory,[25] anti-diabetic,[26] and anti-cancer characteristics [27-31]. The FDA has approved several sulfonamide derivatives as anti-cancer agents, which is noteworthy. For example, Belinostat, an inhibitor of histone deacetylase, has been approved as the third treatment for T-cell lymphoma (cancer of lymph nodes), following Vorinostat and Romidepsin. ABT 199, a Bcl-2 inhibitor, is officially authorized to treat patients with chronic lymphocytic leukemia. Amsacrine has been approved to treat malignant lymphomas and acute leukemias by intercalating tumor DNA and inhibiting topoisomerase-II.[21]
Based on findings published in the medical and pharmaceutical journals, there has been a significant increase in interest in sulfonamide-quinoline hybrids as anti-cancer medicines. [32-40]
Sulfonamides and quinoline derivatives are two pharmacologically relevant scaffolds that have exhibited significant potential in anticancer drug development. Sulfonamides are already known to be inhibitors of carbonic anhydrase IX (CA IX), [41][42][43]an overexpressed isoenzyme in hypoxic tumor microenvironments and involved in tumor survival and metastasis by regulation of extracellular pH [44][45]. Though CA IX expression is low in regular tissues, it is upregulated in bladder, renal, breast, cervical, lung, and colon cancers, making it a potential therapeutic target [38][46]. Quinoline-based derivatives, however, have strong anticancer activity by multiple mechanisms, such as inhibition of Aurora kinases A and B [40], which are serine/threonine kinases playing roles in mitotic cell cycle progression, segregation of chromosomes, and cell cycle regulation. Overexpression of these kinases correlates with numerous malignancies and unfavorable clinical outcomes. [47-52] AURKA is amplified in breast and lung cancers, and AURKB is overexpressed in prostate, NSCLC, glioblastoma, and breast cancer.[53]
Since CA IX and Aurora kinases play complementary and antagonistic roles in cancer biology, the development of hybrid molecules with dual targeting potential towards both pathways is a new and interesting therapeutic approach. Herein, we optimized a set of 32 new sulphonamide-quinoline derivatives through the molecular hybridization strategy. The molecules were structurally optimized by joining sulphonamide and quinoline units via different amino linkers to investigate conformational flexibility and maximize dual-target interaction. This study provides a foundation for the discovery of multi-targeted anticancer drugs and opens up a foundation for additional synthesis, biological testing, and SAR investigations.
Materials and Methods
Design of proposed compounds
The molecular hybridization technique is widely utilized in drug design and discovery, combining various bioactive components to create novel hybrids with enhanced activity.[54] These findings about sulfonamides and quinolines as anticancer medicines prompted the creation of novel sulphonamide-quinoline derivatives as anti-cancer agents. In this research work, we designed 32 novel sulphonamide-quinoline derivatives incorporating different amino linkers [x] [Fig.1] with anticancer potential using the molecular hybridization technique. This linkage is crucial for maintaining the structural integrity. Quinoline moiety is kept rigid, and the substituents attached to the sulphonamide group are denoted by ‘R’. These groups can be varied to modulate the physicochemical properties and biological activity of the hybrids.
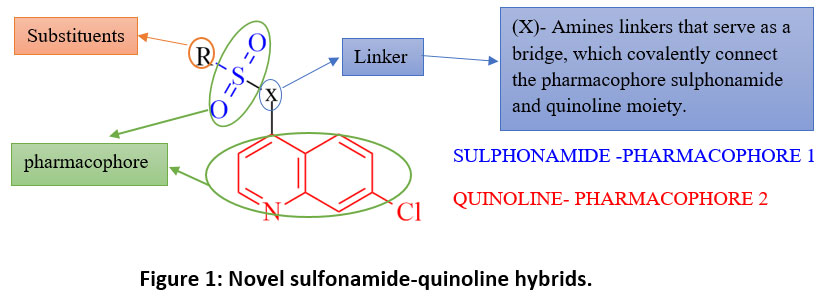 |
Figure 1: Novel sulfonamide-quinoline hybrids.Click here to View Figure |
Table 1: List of 32 Sulfonamide-quinoline derivatives:
| Sr. No. | COMPOUND | R | X |
| 1. | A1 | -phenyl | -1,2-diaminopropane |
| 2. | A2 | -phenyl | -1,3-diaminopropane |
| 3. | A3 | -phenyl | -1,4-diaminobutane |
| 4. | A4 | -phenyl | -benzene 1,2-diamine |
| 5. | A5 | -phenyl | -benzene 1,3-diamine |
| 6. | A6 | -phenyl | -benzene 1,4-diamine |
| 7. | A7 | -phenyl | -Hydrazine |
| 8. | A8 | -phenyl | -piperazine |
| 9. | B1 | -p-toluene | -1,2-diaminopropane |
| 10. | B2 | -p-toluene | -1,3-diaminopropane |
| 11. | B3 | -p-toluene | -1,4-diaminobutane |
| 12. | B4 | -p-toluene | -benzene 1,2-diamine |
| 13. | B5 | -p-toluene | -benzene 1,3-diamine |
| 14. | B6 | -p-toluene | -benzene 1,4-diamine |
| 15. | B7 | -p-toluene | -Hydrazine |
| 16. | B8 | -p-toluene | -piperazine |
| 17. | C1 | -phenyl methane | -1,2-diaminopropane |
| 18. | C2 | -phenyl methane | -1,3-diaminopropane |
| 19. | C3 | -phenyl methane | -1,4-diaminobutane |
| 20. | C4 | -phenyl methane | -benzene 1,2-diamine |
| 21. | C5 | -phenyl methane | -benzene 1,3-diamine |
| 22. | C6 | -phenyl methane | -benzene 1,4-diamine |
| 23. | C7 | -phenyl methane | -Hydrazine |
| 24. | C8 | -phenyl methane | -piperazine |
| 25. | D1 | -4-chlorophenyl | -1,2-diaminopropane |
| 26. | D2 | -4-chlorophenyl | -1,3-diaminopropane |
| 27. | D3 | -4-chlorophenyl | -1,4-diaminobutane |
| 28. | D4 | -4-chlorophenyl | -benzene 1,2-diamine |
| 29. | D5 | -4-chlorophenyl | -benzene 1,3-diamine |
| 30. | D6 | -4-chlorophenyl | -benzene 1,4-diamine |
| 31. | D7 | -4-chlorophenyl | -Hydrazine |
| 32. | D8 | -4-chlorophenyl | -piperazine |
2D structures of designed derivatives of series A, B, C, and D
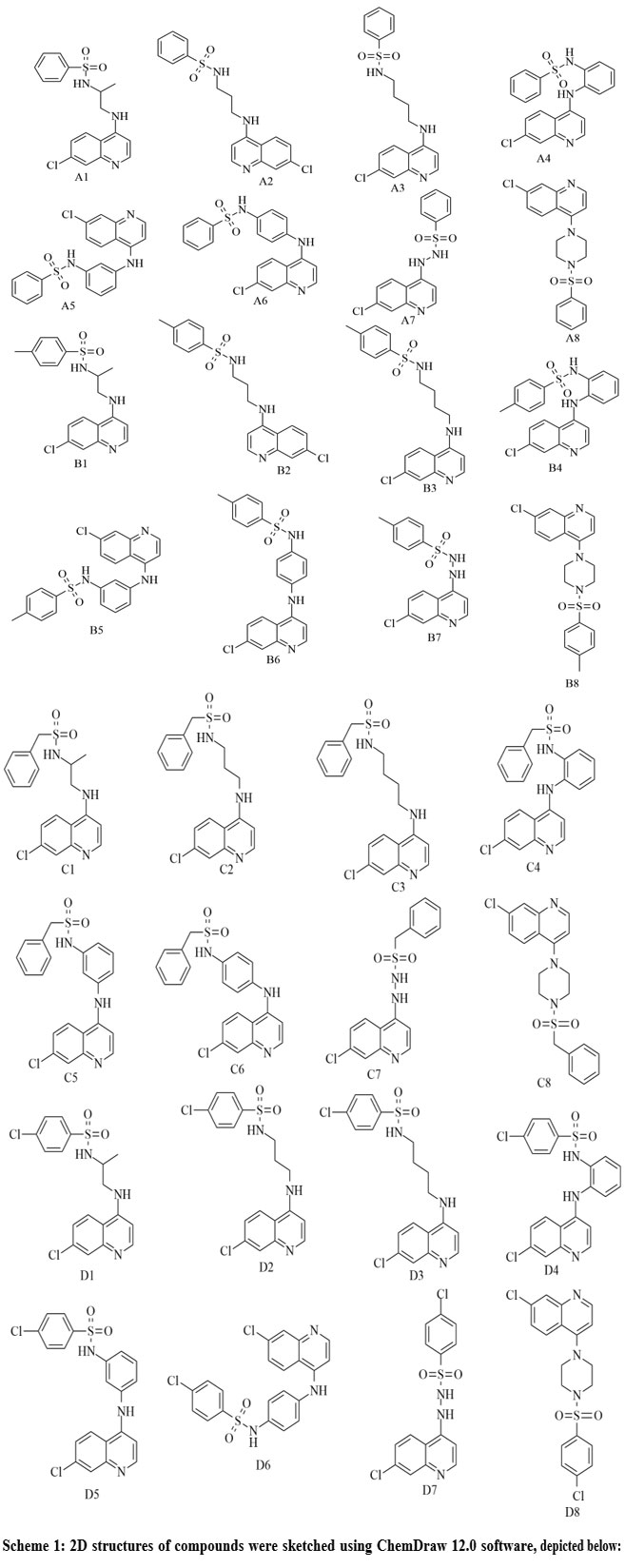 |
Scheme 1: 2D structures of compounds were sketched using ChemDraw 12.0 software, depicted below:Click here to View Scheme |
Identification of potent compounds by docking studies
2D- structures of designed compounds were drawn in ChemDraw 12 software & Energy Minimization was done using Chem3D Pro 12.0, and minimized structures were saved in mol format, and further docking was performed stepwise to identify potent compounds.
PDB ID Validation
The molecular structures for target CAIX, AURKA, and AURKB were retrieved in pdb. format from the protein database with PDB IDs 5FL6, 3K5U, and 4AF3, respectively. These PDB IDs were optimized based on the normal procedure. Validation was done to confirm the docking procedure.
Docking of designed quinoline-sulphonamide hybrids
Docking studies were conducted for designed molecules (Table 1). Docking studies were performed with GOLD 5.3.0 (Cambridge Crystallographic Data Centre, Cambridge, UK). Goldscore is used as a scoring function based on binding affinity. The cavity was only 10Å from the molecular attachment affinity. For every isomer, docking was conducted at least three times, and each position had a corresponding Goldscore fitness function ranking. Top-score compounds were selected for further studies.
Results and Discussion
PDB ID validation
The RMSD value was obtained for CA IX (5FL6): 0.2Å (as shown in Fig.2), AURKA(3K5U): 0.2Å (as shown in Fig.3), AURKB(4AF3): 0.7Å (as shown in Fig.4).
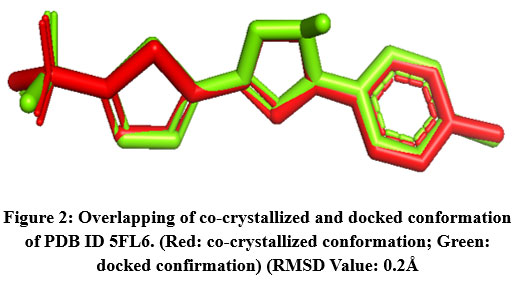 |
Figure 2: Overlapping of co-crystallized and docked conformation of PDB ID 5FL6. (Red: co-crystallized conformation; Green: docked confirmation) (RMSD Value: 0.2Å.Click here to View Figure |
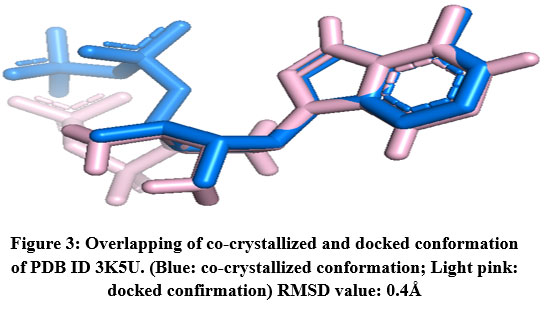 |
Figure 3: Overlapping of co-crystallized and docked conformation of PDB ID 3K5U. (Blue: co-crystallized conformation; Light pink: docked confirmation) RMSD value: 0.4Å.Click here to View Figure |
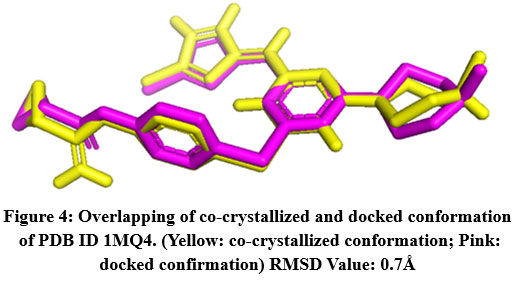 |
Figure 4: Overlapping of co-crystallized and docked conformation of PDB ID 1MQ4. (Yellow: co-crystallized conformation; Pink: docked confirmation) RMSD Value: 0.7Å.Click here to View Figure |
Docking scores of docked compounds
The docking score of the docked compounds is shown in Table 2
Table 2: Docking scores (kcal/mol) of Derivatives
| Compound | Target | ||
| CA IX | AURKA | AURKB | |
| A1 | 59.00 | 61.28 | 62.53 |
| A2 | 56.94 | 73.34 | 67.53 |
| A3 | 57.72 | 66.23 | 66.63 |
| A4 | 55.98 | 55.99 | 61.10 |
| A5 | 61.11 | 71.21 | 67.87 |
| A6 | 53.48 | 74.35 | 67.48 |
| A7 | 58.67 | 56.31 | 57.87 |
| A8 | 54.43 | 58.44 | 58.93 |
| B1 | 60.24 | 65.27 | 65.53 |
| B2 | 58.06 | 71.82 | 66.36 |
| B3 | 61.21 | 73.21 | 66.87 |
| B4 | 55.62 | 57.45 | 56.35 |
| B5 | 64.07 | 72.97 | 69.17 |
| B6 | 54.39 | 74.24 | 69.34 |
| B7 | 60.06 | 58.85 | 60.69 |
| B8 | 56.18 | 62.31 | 60.79 |
| C1 | 60.07 | 61.45 | 69.56 |
| C2 | 56.97 | 66.78 | 66.45 |
| C3 | 60.87 | 68.96 | 71.43 |
| C4 | 60.72 | 60.55 | 63.88 |
| C5 | 61.60 | 68.23 | 71.24 |
| C6 | 56.85 | 66.73 | 71.05 |
| C7 | 56.87 | 63.30 | 71.05 |
| C8 | 48.65 | 62.01 | 70.92 |
| D1 | 65.20 | 65.34 | 65.84 |
| D2 | 62.98 | 72.09 | 69.07 |
| D3 | 61.13 | 72.48 | 67.01 |
| D4 | 58.05 | 60.27 | 64.36 |
| D5 | 59.49 | 64.42 | 63.43 |
| D6 | 58.70 | 73.30 | 68.67 |
| D7 | 58.66 | 57.18 | 61.06 |
| D8 | 55.40 | 63.54 | 69.29 |
| SLC-011* | 62.95 | 53.48 | 53.44 |
| Sitamaquine* | 59.29 | 63.35 | 59.66 |
Sitamaquine*, SLC-011*- Reference drug
B5, B3, C5, C3, A5, and D2 showed overall good Fitness scores for all three enzymes, CAIX, AURKA, and AURKB.
A2, A6, B6, D3, and D6 showed good fitness scores for enzymes AURKA and AURKB.
Designed derivatives also showed better fitness scores than the reference drugs- SLC-011 and Sitamaquine.
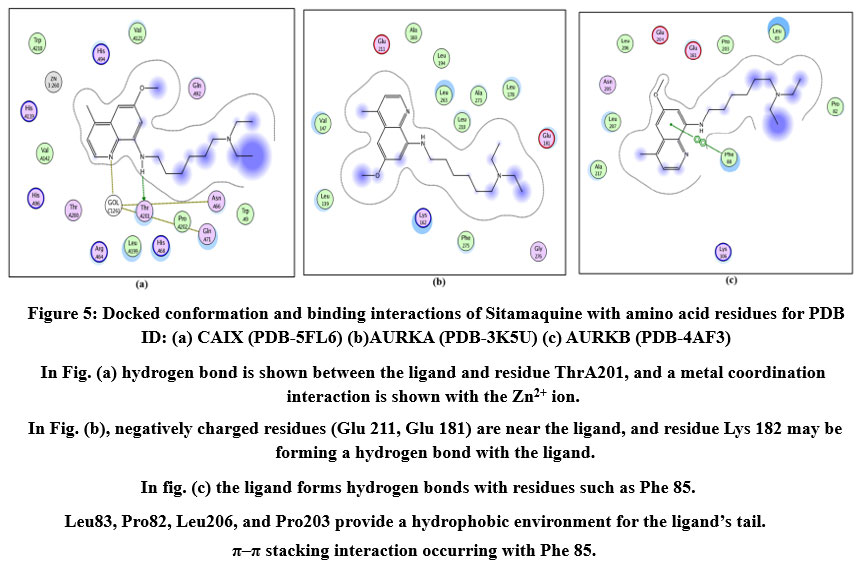 |
Figure 5: Docked conformation and binding interactions of Sitamaquine with amino acid residues for PDB ID: (a) CAIX (PDB-5FL6) (b)AURKA (PDB-3K5U) (c) AURKB (PDB-4AF3).Click here to View Figure |
2D Images of the interaction of Reference drugs
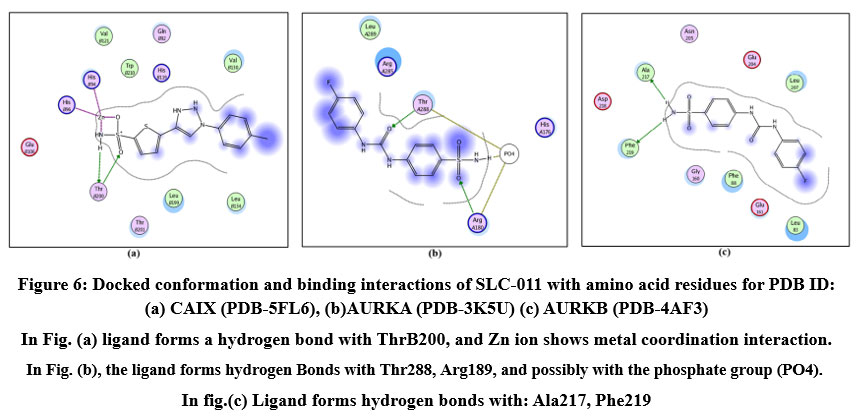 |
Figure 6: Docked conformation and binding interactions of SLC-011 with amino acid residues for PDB ID: (a) CAIX (PDB-5FL6), (b)AURKA (PDB-3K5U) (c) AURKB (PDB-4AF3).Click here to View Figure |
2D interaction images of best-docked compounds
2D interaction images of best-docked compounds in all three target proteins, CA IX, AURKA, and AURKB, respectively, are provided below:
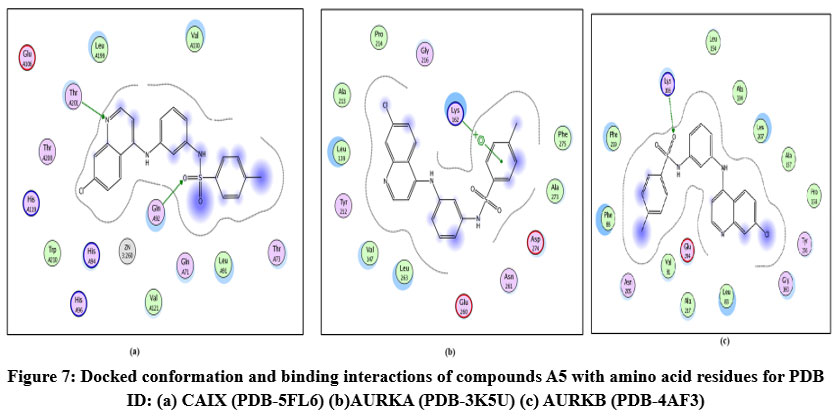 |
Figure 7: Docked conformation and binding interactions of compounds A5 with amino acid residues for PDB ID: (a) CAIX (PDB-5FL6) (b)AURKA (PDB-3K5U) (c) AURKB (PDB-4AF3).Click here to View Figure |
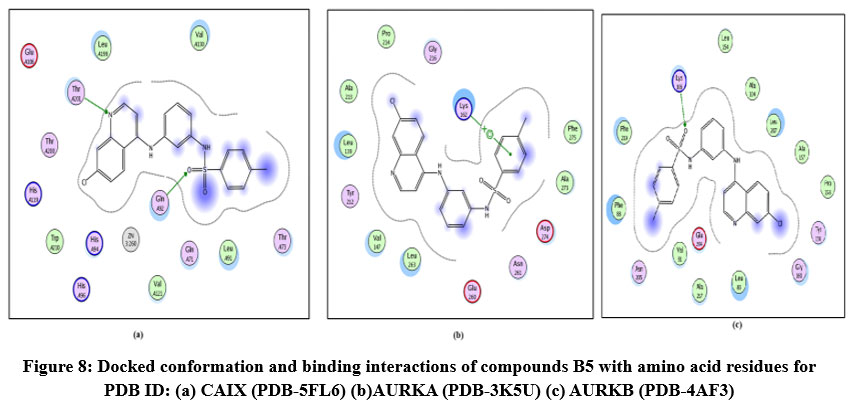 |
Figure 8: Docked conformation and binding interactions of compounds B5 with amino acid residues for PDB ID: (a) CAIX (PDB-5FL6) (b)AURKA (PDB-3K5U) (c) AURKB (PDB-4AF3).Click here to View Figure |
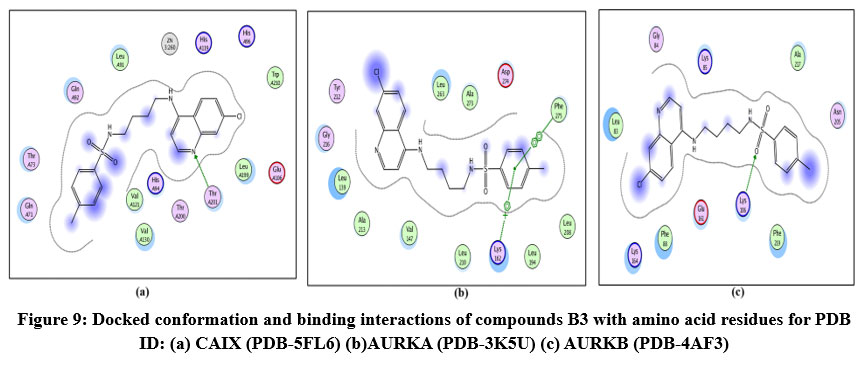 |
Figure 9: Docked conformation and binding interactions of compounds B3 with amino acid residues for PDB ID: (a) CAIX (PDB-5FL6) (b)AURKA (PDB-3K5U) (c) AURKB (PDB-4AF3).Click here to View Figure |
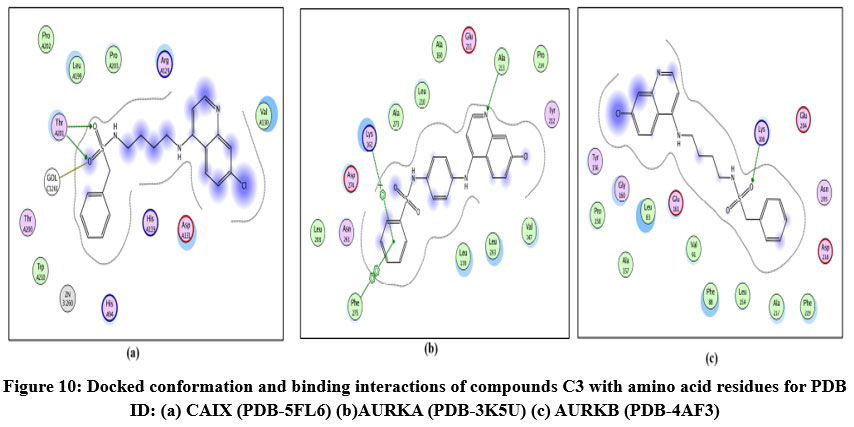 |
Figure 10: Docked conformation and binding interactions of compounds C3 with amino acid residues for PDB ID: (a) CAIX (PDB-5FL6) (b)AURKA (PDB-3K5U) (c) AURKB (PDB-4AF3).Click here to View Figure |
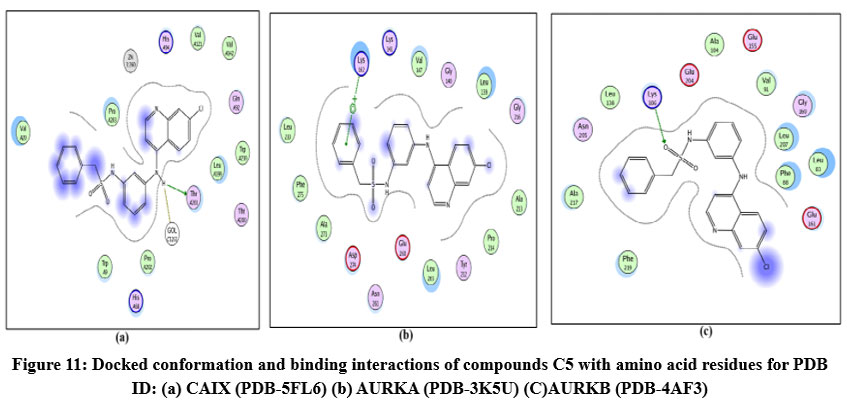 |
Figure 11: Docked conformation and binding interactions of compounds C5 with amino acid residues for PDB ID: (a) CAIX (PDB-5FL6) (b) AURKA (PDB-3K5U) (C)AURKB (PDB-4AF3).Click here to View Figure |
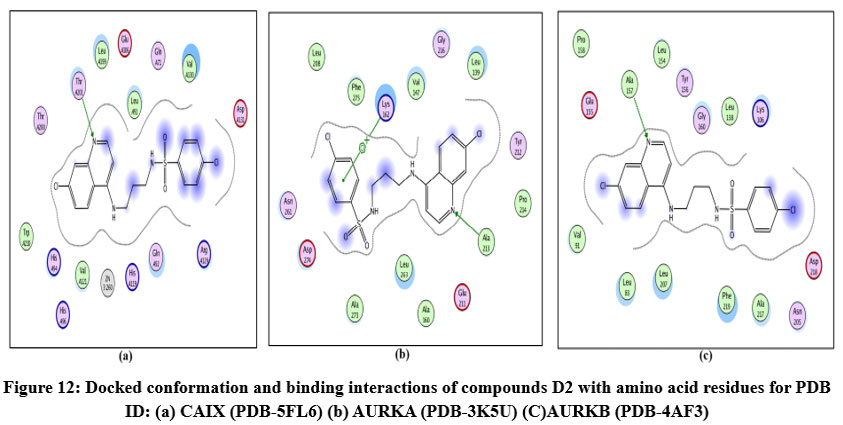 |
Figure 12: Docked conformation and binding interactions of compounds D2 with amino acid residues for PDB ID: (a) CAIX (PDB-5FL6) (b) AURKA (PDB-3K5U) (C)AURKB (PDB-4AF3).Click here to View Fgure |
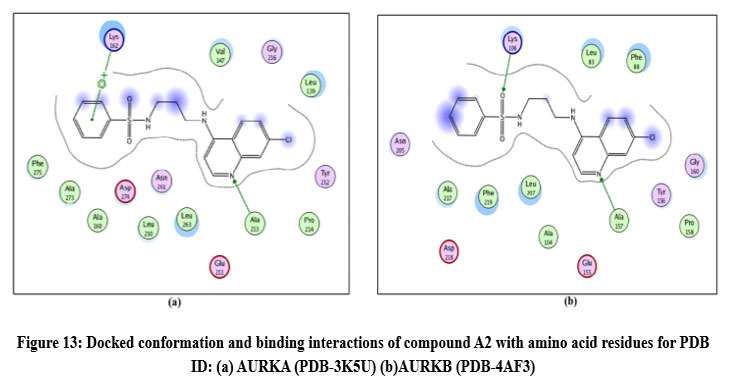 |
Figure 13: Docked conformation and binding interactions of compound A2 with amino acid residues for PDB ID: (a) AURKA (PDB-3K5U) (b)AURKB (PDB-4AF3).Click here to View Figure |
Panel (a): Interactions with CA IX
Compound A5, B5, B3, C3, C5 and D2 showing interaction with residue ThrA201, Quinoline moiety of these compounds forming a hydrogen bond with ThrA201, except for C5 and C3, in which the linker and Sulphonamide moiety form a hydrogen bond with ThrA201, respectively.
Zn²⁺ Ion Coordination (ZN at 3.260) suggests additional metal coordination interaction.
Hydrophobic Environment: Surrounded by Leu, Val, Trp, His — stabilizing the ligand in the pocket.
Panel (b): Interactions with AURKA
Compound A5, B5, B3, C3, C5 and D2 showing arene-cation interaction with residue Lys162 forming hydrogen bonds. In addition to this, B3 and C3 show an arene-arene interaction with residue Phe275, forming a hydrogen bond.
Compound D2 also forms a hydrogen bond with residue ALA213.
Charged residues nearby: Asp274, Lys162 (positively charged), help stabilize the ligand
Panel (c): Interactions with AURKB
Compound A5, B5, B3, C3, C5 and D2 forming single hydrogen bond with residue Lys106.
Hydrophobic and polar residues:
Hydrophobic: Phe219, Leu83, Leu207
Polar: Gly84, Asn205, Ala227.
2D interaction images of best-docked compounds with AURKA and AURKB are provided below:
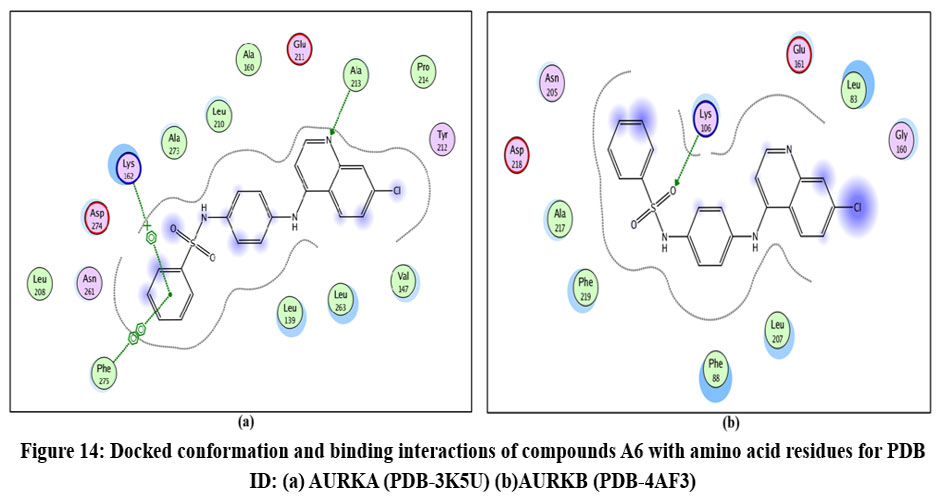 |
Figure 14: Docked conformation and binding interactions of compounds A6 with amino acid residues for PDB ID: (a) AURKA (PDB-3K5U) (b)AURKB (PDB-4AF3).Click here to View Figure |
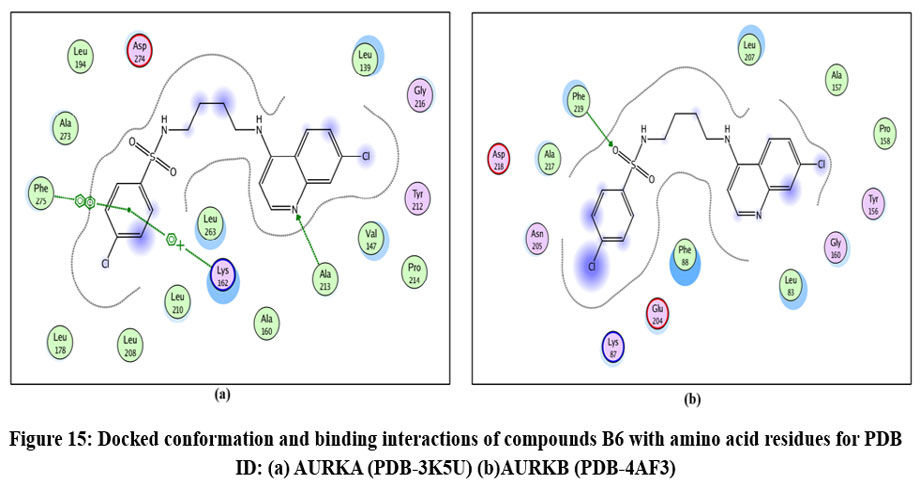 |
Figure 15: Docked conformation and binding interactions of compounds B6 with amino acid residues for PDB ID: (a) AURKA (PDB-3K5U) (b)AURKB (PDB-4AF3). Click here to View Figure |
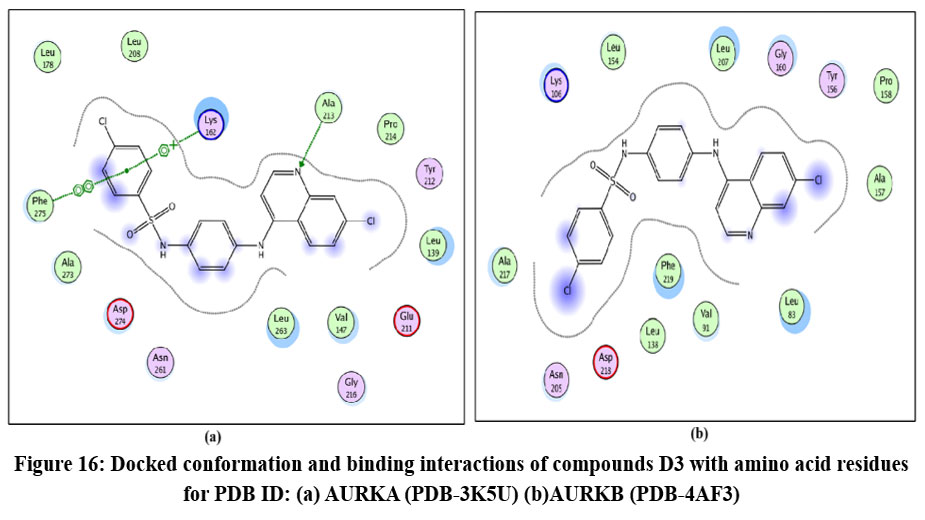 |
Figure 16: Docked conformation and binding interactions of compounds D3 with amino acid residues for PDB ID: (a) AURKA (PDB-3K5U) (b)AURKB (PDB-4AF3).Click here to View table |
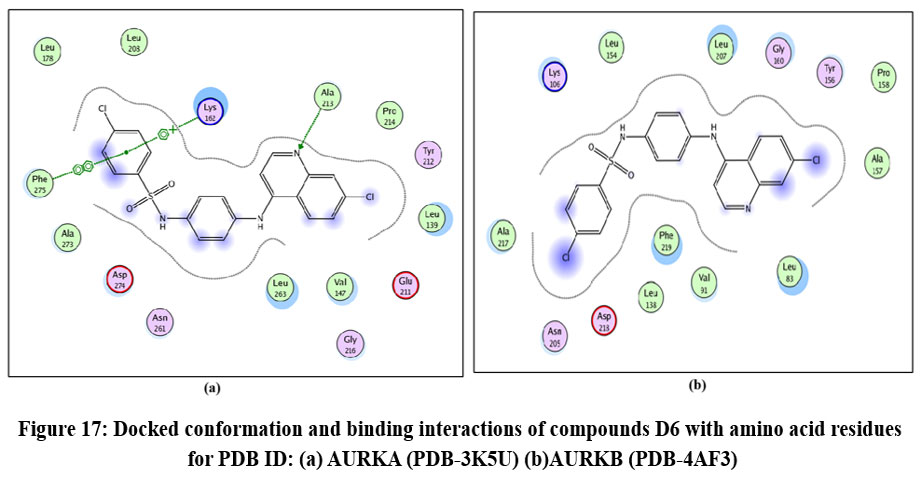 |
Figure 17: Docked conformation and binding interactions of compounds D6 with amino acid residues for PDB ID: (a) AURKA (PDB-3K5U) (b)AURKB (PDB-4AF3). Click here to View Figure |
Panel (b): Interactions with AURKA enzyme
Compounds A2, A6, B6, D6, D3 showed interactions with residue Lys162, Phe275 forming hydrogen bonds.
Panel (c): Interactions with AURKB enzyme
Compound A2 and A6 formed a hydrogen bond with Lys 106, and B6 showed interaction with residue Phe219, forming a hydrogen bond.
Important amino acids involved in the interaction of the drug-receptor complex
Table 3: Type of interactions and amino acid residues involved for compounds
| Panel | Target Enzyme | Compounds | Amino acid Residues Involved | Interaction Type | Additional Notes |
| (a) | CA IX | A5, B5, B3, C3, C5, D2 | ThrA201 | H-bond (via Quinoline moiety) | C5 (via linker), C3 (via sulphonamide) |
| (a) | CA IX | Zn²⁺ at 3.260 Å | Metal coordination | Suggests strong binding | |
| (a) | CA IX | Leu, Val, Trp, His | Hydrophobic interactions | Pocket stabilization | |
| (b) | AURKA | A5, B5, B3, C3, C5, D2 | Lys162 | Arene-cation + H-bond | Key interaction site |
| (b) | AURKA | Phe275 | Arene-arene + H-bond | Observed in B3, C3 | |
| (b) | AURKA | Ala213 | H-bond | Observed in D2 | |
| (b) | AURKA | Asp274, Lys162 | Electrostatic stabilizing | Positively charged pocket | |
| (b) | AURKA | A2, A6, B6, D6, D3 | Lys162, Phe275 | H-bonds | Alternate compound set |
| (c) | AURKB | A5, B5, B3, C3, C5, D2 | Lys106 | H-bond | Common interaction site |
| (c) | AURKB | Phe219, Leu83, Leu207 | Hydrophobic | Ligand stabilization | |
| (c) | AURKB | Gly84, Asn205, Ala227 | Polar interactions | Enhance binding | |
| (c) | AURKB | A2, A6 | Lys106 | H-bond | Confirmed interaction |
| (c) | AURKB | B6 | Phe219 | H-bond | Additional interaction |
Study profile of designed derivatives
Physicochemical properties
Key physicochemical characteristics related to bioavailability and drug-likeness were examined in 32 designed compounds (A1–D8) as well as the reference molecule SLC-011. These include molecular weight (MW), number of rotatable bonds, hydrogen bond donors (HBD), and topological polar surface area (TPSA). Most derivatives show moderate molecular weights (MW: 333.79–444.33 Da) and acceptable topological polar surface area (TPSA: ~61.89–79.47 Ų), suggesting good oral bioavailability.As stated in Table 4, all of the proposed derivatives adhere to the Lipinski rule of five, which indicates that excellent absorption is more likely to occur when there are no more than (i) five H-bond donors, (ii) ten H-bond acceptors, (iii) the molecular weight (MW) <500 dalton, and (iv) a computed Log P (cLogP) less than five.[55]As shown in Table 5, Most molecules have LogP values between 2 and 6, indicating moderate lipophilicity, which is ideal for drug-likeness.
Solubility prediction
Solubility predictions using ESOL, Ali, and Silicos-IT methods showed variability in aqueous solubility and classification (Table 6). The majority of compounds are classified as moderately soluble (MS) or poorly soluble (PS), except for SLC-011 which is soluble (S).
Pharmacokinetic parameters
Pharmacokinetic parameters are indicated in Table 7. The faster that it is broken down and digested in the body, the higher the GI absorption. Most of the molecules have high GI absorption, except B4, D4, D5, and D6. This is, therefore, good oral bioavailability. Only A8, B8, C8, D8, and Sitamaquine can cross the BBB and hence are possible candidates for the CNS-related treatments. B4, D4, D5, and D6 require either prodrugs or formulation changes due to low GI absorption.
Table 4: Physicochemical properties of designed derivatives of series A, B, C, and D
| Molecule | Formula | MW | Heavy atoms | Aromatic heavy atoms | Fraction Csp3 | Rotatable bonds | H-bond acceptors | H-bond donors | MR | TPSA |
| A1 | C18H18ClN3O2S | 375.87 | 25 | 16 | 0.17 | 6 | 4 | 2 | 101.09 | 79.47 |
| A2 | C18H18ClN3O2S | 375.87 | 25 | 16 | 0.17 | 7 | 4 | 2 | 101.09 | 79.47 |
| A3 | C19H20ClN3O2S | 389.9 | 26 | 16 | 0.21 | 8 | 4 | 2 | 105.9 | 79.47 |
| A4 | C21H16ClN3O2S | 409.89 | 28 | 22 | 0 | 5 | 3 | 2 | 113.41 | 79.47 |
| A5 | C21H16ClN3O2S | 409.89 | 28 | 22 | 0 | 5 | 3 | 2 | 113.41 | 79.47 |
| A6 | C21H16ClN3O2S | 409.89 | 28 | 22 | 0 | 5 | 3 | 2 | 113.41 | 79.47 |
| A7 | C15H12ClN3O2S | 333.79 | 22 | 16 | 0 | 4 | 4 | 2 | 86.67 | 79.47 |
| A8 | C19H18ClN3O2S | 387.88 | 26 | 16 | 0.21 | 3 | 4 | 0 | 110.82 | 61.89 |
| B1 | C19H20ClN3O2S | 389.9 | 26 | 16 | 0.21 | 6 | 4 | 2 | 106.06 | 79.47 |
| B2 | C19H20ClN3O2S | 389.9 | 26 | 16 | 0.21 | 7 | 4 | 2 | 106.06 | 79.47 |
| B3 | C20H22ClN3O2S | 403.93 | 27 | 16 | 0.25 | 8 | 4 | 2 | 110.86 | 79.47 |
| B4 | C22H18ClN3O2S | 423.92 | 29 | 22 | 0.05 | 5 | 3 | 2 | 118.38 | 79.47 |
| B5 | C22H18ClN3O2S | 423.92 | 29 | 22 | 0.05 | 5 | 3 | 2 | 118.38 | 79.47 |
| B6 | C22H18ClN3O2S | 423.92 | 29 | 22 | 0.05 | 5 | 3 | 2 | 118.38 | 79.47 |
| B7 | C16H14ClN3O2S | 347.82 | 23 | 16 | 0.06 | 4 | 4 | 2 | 91.63 | 79.47 |
| B8 | C20H20ClN3O2S | 401.91 | 27 | 16 | 0.25 | 3 | 4 | 0 | 115.79 | 61.89 |
| C1 | C19H20ClN3O2S | 389.9 | 26 | 16 | 0.21 | 7 | 4 | 2 | 106.73 | 79.47 |
| C2 | C19H20ClN3O2S | 389.9 | 26 | 16 | 0.21 | 8 | 4 | 2 | 106.73 | 79.47 |
| C3 | C20H22ClN3O2S | 403.93 | 27 | 16 | 0.25 | 9 | 4 | 2 | 111.54 | 79.47 |
| C4 | C22H18ClN3O2S | 423.92 | 29 | 22 | 0.05 | 6 | 3 | 2 | 119.05 | 79.47 |
| C5 | C22H18ClN3O2S | 423.92 | 29 | 22 | 0.05 | 6 | 3 | 2 | 119.05 | 79.47 |
| C6 | C22H18ClN3O2S | 423.92 | 29 | 22 | 0.05 | 6 | 3 | 2 | 119.05 | 79.47 |
| C7 | C16H14ClN3O2S | 347.82 | 23 | 16 | 0.06 | 5 | 4 | 2 | 92.31 | 79.47 |
| C8 | C20H20ClN3O2S | 401.91 | 27 | 16 | 0.25 | 4 | 4 | 0 | 116.46 | 61.89 |
| D1 | C18H17Cl2N3O2S | 410.32 | 26 | 16 | 0.17 | 6 | 4 | 2 | 106.1 | 79.47 |
| D2 | C18H17Cl2N3O2S | 410.32 | 26 | 16 | 0.17 | 7 | 4 | 2 | 106.1 | 79.47 |
| D3 | C19H19Cl2N3O2S | 424.34 | 27 | 16 | 0.21 | 8 | 4 | 2 | 110.91 | 79.47 |
| D4 | C21H15Cl2N3O2S | 444.33 | 29 | 22 | 0 | 5 | 3 | 2 | 118.42 | 79.47 |
| D5 | C21H15Cl2N3O2S | 444.33 | 29 | 22 | 0 | 5 | 3 | 2 | 118.42 | 79.47 |
| D6 | C21H15Cl2N3O2S | 444.33 | 29 | 22 | 0 | 5 | 3 | 2 | 118.42 | 79.47 |
| D7 | C15H11Cl2N3O2S | 368.24 | 23 | 16 | 0 | 4 | 4 | 2 | 91.68 | 79.47 |
| D8 | C19H17Cl2N3O2S | 422.33 | 27 | 16 | 0.21 | 3 | 4 | 0 | 115.83 | 61.89 |
| SLC-011 | C13H12FN3O3S | 309.32 | 21 | 12 | 0 | 5 | 5 | 3 | 76.12 | 109.67 |
Table 5: Lipophilicity of designed derivatives of series A, B, C and D
| Molecule | iLOGP | XLOGP3 | WLOGP | MLOGP | Silicos-IT Log P |
| A1 | 2.15 | 3.9 | 4.56 | 2.22 | 2.8 |
| A2 | 2.82 | 4.98 | 4.56 | 2.22 | 2.97 |
| A3 | 3.47 | 4.18 | 4.95 | 2.45 | 3.36 |
| A4 | 2.73 | 5.49 | 6.32 | 3.21 | 3.39 |
| A5 | 2.92 | 5.49 | 6.32 | 3.21 | 3.39 |
| A6 | 2.82 | 5.49 | 6.32 | 3.21 | 3.39 |
| A7 | 1.86 | 4.27 | 4.08 | 2.32 | 1.84 |
| A8 | 3.01 | 3.56 | 3.72 | 2.45 | 2.42 |
| B1 | 2.63 | 4.26 | 4.87 | 2.45 | 3.32 |
| B2 | 2.87 | 4.74 | 4.87 | 2.45 | 3.49 |
| B3 | 3.13 | 4.54 | 5.26 | 2.67 | 3.89 |
| B4 | 3.17 | 5.86 | 6.63 | 3.43 | 3.91 |
| B5 | 3.18 | 5.86 | 6.63 | 3.43 | 3.91 |
| B6 | 2.97 | 5.86 | 6.63 | 3.43 | 3.91 |
| B7 | 2.92 | 4.63 | 4.39 | 2.56 | 2.34 |
| B8 | 3.28 | 3.92 | 4.03 | 2.67 | 2.93 |
| C1 | 2.38 | 3.83 | 4.55 | 2.18 | 3.19 |
| C2 | 2.39 | 4.31 | 4.55 | 2.18 | 3.36 |
| C3 | 2.65 | 4.11 | 4.94 | 2.4 | 3.75 |
| C4 | 2.33 | 4.82 | 6.31 | 3.16 | 3.78 |
| C5 | 2.61 | 5.43 | 6.31 | 3.16 | 3.78 |
| C6 | 2.86 | 5.43 | 6.31 | 3.16 | 3.78 |
| C7 | 2.23 | 4.2 | 4.07 | 2.29 | 2.21 |
| C8 | 2.74 | 3.49 | 3.71 | 2.4 | 2.8 |
| D1 | 2.72 | 5.13 | 5.21 | 2.72 | 3.45 |
| D2 | 2.7 | 5.61 | 5.21 | 2.72 | 3.62 |
| D3 | 3.2 | 5.41 | 5.6 | 2.94 | 4.01 |
| D4 | 3.01 | 6.12 | 6.98 | 3.7 | 4.03 |
| D5 | 2.89 | 6.12 | 6.98 | 3.7 | 4.03 |
| D6 | 3.05 | 6.12 | 6.98 | 3.7 | 4.03 |
| D7 | 2.18 | 4.89 | 4.74 | 2.83 | 2.48 |
| D8 | 3.29 | 4.19 | 4.37 | 2.94 | 3.05 |
| SLC-011 | 1.56 | 1.45 | 3.24 | 1.78 | 0.34 |
| Sitamaquine | 4.43 | 4.71 | 4.68 | 2.8 | 4.87 |
Table 6: Water solubility of designed derivatives of series A, B, C and D
| Molecule | ESOL | Ali | Silicos-IT | ||||||
| Log S | Solubility(mg/ml) | Class | Log S | Solubility(mg/ml) | Class | Log S | Solubility | class | |
| A1 | -4.71 | 7.41E-03 | MS | -5.27 | 2.03E-03 | MS | -7.64 | 2.27E-08 | PS |
| A2 | -5.32 | 1.80E-03 | MS | -6.39 | 1.54E-04 | PS | -8.02 | 9.59E-09 | PS |
| A3 | -4.82 | 5.93E-03 | MS | -5.56 | 1.08E-03 | MS | -8.41 | 3.87E-09 | PS |
| A4 | -6.09 | 3.32E-04 | PS | -6.92 | 4.96E-05 | PS | -9.29 | 5.11E-10 | PS |
| A5 | -6.09 | 3.32E-04 | PS | -6.92 | 4.96E-05 | PS | -9.29 | 5.11E-10 | PS |
| A6 | -6.09 | 3.32E-04 | PS | -6.92 | 4.96E-05 | PS | -9.29 | 5.11E-10 | PS |
| A7 | -4.87 | 4.46E-03 | MS | -5.65 | 7.45E-04 | MS | -6.83 | 1.47E-07 | PS |
| A8 | -4.75 | 6.98E-03 | MS | -4.55 | 1.11E-02 | MS | -6.47 | 3.37E-07 | PS |
| B1 | -5 | 3.89E-03 | MS | -5.64 | 8.92E-04 | MS | -8.02 | 9.55E-09 | PS |
| B2 | -5.24 | 2.26E-03 | MS | -6.14 | 2.83E-04 | PS | -8.39 | 4.04E-09 | PS |
| B3 | -5.12 | 3.10E-03 | MS | -5.93 | 4.73E-04 | MS | -8.79 | 1.63E-09 | PS |
| B4 | -6.39 | 1.72E-04 | PS | -7.3 | 2.12E-05 | PS | -9.67 | 2.15E-10 | PS |
| B5 | -6.39 | 1.72E-04 | PS | -7.3 | 2.12E-05 | PS | -9.67 | 2.15E-10 | PS |
| B6 | -6.39 | 1.72E-04 | PS | -7.3 | 2.12E-05 | PS | -9.67 | 2.15E-10 | PS |
| B7 | -5.16 | 2.38E-03 | MS | -6.02 | 3.29E-04 | PS | -7.21 | 6.15E-08 | PS |
| B8 | -5.04 | 3.65E-03 | MS | -4.92 | 4.85E-03 | MS | -6.85 | 1.42E-07 | PS |
| C1 | -4.66 | 8.46E-03 | MS | -5.19 | 2.49E-03 | MS | -8.04 | 9.16E-09 | PS |
| C2 | -4.9 | 4.91E-03 | MS | -5.69 | 7.91E-04 | MS | -8.41 | 3.87E-09 | PS |
| C3 | -4.78 | 6.73E-03 | MS | -5.49 | 1.32E-03 | MS | -8.8 | 1.57E-09 | PS |
| C4 | -5.67 | 9.06E-04 | MS | -6.22 | 2.54E-04 | PS | -9.68 | 2.07E-10 | PS |
| C5 | -6.05 | 3.74E-04 | PS | -6.85 | 5.92E-05 | PS | -9.68 | 2.07E-10 | PS |
| C6 | -6.05 | 3.74E-04 | PS | -6.85 | 5.92E-05 | PS | -9.68 | 2.07E-10 | PS |
| C7 | -4.83 | 5.18E-03 | MS | -5.58 | 9.18E-04 | MS | -7.23 | 5.90E-08 | PS |
| C8 | -4.71 | 7.93E-03 | MS | -4.47 | 1.35E-02 | MS | -6.87 | 1.36E-07 | PS |
| D1 | -5.68 | 8.67E-04 | MS | -6.54 | 1.17E-04 | PS | -8.23 | 5.86E-09 | PS |
| D2 | -5.91 | 5.03E-04 | MS | -7.04 | 3.73E-05 | PS | -8.61 | 2.48E-09 | PS |
| D3 | -5.79 | 6.89E-04 | MS | -6.83 | 6.22E-05 | PS | -9 | 1.01E-09 | PS |
| D4 | -6.68 | 9.24E-05 | PS | -7.57 | 1.19E-05 | PS | -9.88 | 1.33E-10 | PS |
| D5 | -6.68 | 9.24E-05 | PS | -7.57 | 1.19E-05 | PS | -9.88 | 1.33E-10 | PS |
| D6 | -6.68 | 9.24E-05 | PS | -7.57 | 1.19E-05 | PS | -9.88 | 1.33E-10 | PS |
| D7 | -5.45 | 1.29E-03 | MS | -6.29 | 1.87E-04 | PS | -7.43 | 3.75E-08 | PS |
| D8 | -5.34 | 1.94E-03 | MS | -5.2 | 2.67E-03 | MS | -7.06 | 8.75E-08 | PS |
| SLC-011 | -2.76 | 5.32E-01 | S | -3.36 | 1.35E-01 | S | -4.8 | 1.60E-05 | MS |
| Sitamaquine | -4.51 | 1.07E-02 | MS | -5.22 | 2.05E-03 | MS | -7.58 | 2.62E-08 | PS |
Table 7: Pharmacokinetic Parameters of designed derivatives of series A, B, C, and D
| Molecule | GI absorption | BBB permeability | Pgp substrate | CYP1A2 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor |
| A1 | ↑ | – | – | √ | √ | √ | √ | √ |
| A2 | ↑ | – | – | √ | √ | √ | √ | √ |
| A3 | ↑ | – | – | √ | √ | √ | √ | √ |
| A4 | ↑ | – | – | √ | √ | √ | √ | √ |
| A5 | ↑ | – | – | √ | √ | √ | √ | √ |
| A6 | ↑ | – | – | √ | √ | √ | √ | √ |
| A7 | ↑ | – | – | √ | √ | √ | √ | √ |
| A8 | ↑ | – | – | √ | √ | √ | √ | √ |
| B1 | ↑ | – | – | √ | √ | √ | √ | √ |
| B2 | ↑ | – | – | √ | √ | √ | √ | √ |
| B3 | ↑ | – | – | √ | √ | √ | √ | √ |
| B4 | ↓ | – | – | √ | √ | √ | √ | √ |
| B5 | ↑ | – | – | √ | √ | √ | √ | √ |
| B6 | ↑ | – | – | √ | √ | √ | √ | √ |
| B7 | ↑ | – | – | √ | √ | √ | √ | √ |
| B8 | ↑ | √ | – | √ | √ | √ | √ | √ |
| C1 | ↑ | – | – | √ | √ | √ | √ | √ |
| C2 | ↑ | – | – | √ | √ | √ | √ | √ |
| C3 | ↑ | – | – | √ | √ | √ | √ | √ |
| C4 | ↑ | – | – | √ | √ | √ | √ | √ |
| C5 | ↑ | – | – | √ | √ | √ | √ | √ |
| C6 | ↑ | – | – | √ | √ | √ | √ | √ |
| C7 | ↑ | – | – | √ | √ | √ | √ | √ |
| C8 | ↑ | √ | – | √ | √ | √ | √ | √ |
| D1 | ↑ | – | – | √ | √ | √ | √ | √ |
| D2 | ↑ | – | – | √ | √ | √ | √ | √ |
| D3 | ↑ | – | – | √ | √ | √ | √ | √ |
| D4 | ↓ | – | – | √ | √ | √ | √ | √ |
| D5 | ↓ | – | – | √ | √ | √ | √ | √ |
| D6 | ↓ | – | – | √ | √ | √ | √ | √ |
| D7 | ↑ | – | – | √ | √ | √ | √ | √ |
| D8 | ↑ | √ | – | √ | √ | √ | – | √ |
| SLC-011 | ↑ | √ | – | – | – | – | – | – |
| Sitamaquine | ↑ | √ | – | √ | – | – | √ | √ |
↑: maximum absorption, ↓: minimum absorption
Based on the toxicity profile depicted in Table 8, A2, B4, B5, B6, D4, D5, and D6 molecules only demonstrate respiratory toxicity and BBB penetration and are thus relatively safer. These compounds also had less toxicity compared to the standard drug sitamaquine. The derivatives are the most promising, as these compounds do not have neurotoxicity, although they have respiratory toxicity, which requires optimization and makes them potential candidates as anti-cancer agents.
Table 8: Toxicity profile of designed derivatives of series A, B, C and D
| Molecule | Organ toxicity | Toxicity endpoints | ||||||||
| Hepato-toxicity | Neuro-toxicity | Nephro-toxicity | Respiratory toxicity | Cardio-toxicity | Carcino-genicity | Immuno-toxicity | Mutag-enicity | Cytoto-xicity | BBB-barrier | |
| A1 | X | √ | X | √ | X | X | X | X | X | √ |
| A2 | X | X | X | √ | X | X | X | X | X | √ |
| A3 | X | X | X | √ | X | X | X | X | X | √ |
| A4 | X | X | X | √ | X | X | X | X | X | √ |
| A5 | X | X | X | √ | X | X | X | X | X | √ |
| A6 | X | √ | X | √ | X | X | X | X | X | √ |
| A7 | X | √ | X | √ | X | X | √ | X | X | √ |
| A8 | X | √ | X | √ | X | X | √ | X | X | √ |
| B1 | X | √ | X | √ | X | X | X | X | X | √ |
| B2 | X | √ | X | √ | X | X | √ | X | X | √ |
| B3 | X | √ | X | √ | X | X | √ | X | X | √ |
| B4 | X | X | X | √ | X | X | X | X | X | √ |
| B5 | X | X | X | √ | X | X | X | X | X | √ |
| B6 | X | X | X | √ | X | X | X | X | X | √ |
| B7 | X | X | X | √ | X | √ | X | X | X | √ |
| B8 | X | √ | X | √ | X | X | X | X | X | √ |
| C1 | X | √ | X | √ | X | X | √ | X | X | √ |
| C2 | X | √ | X | √ | X | X | √ | √ | X | √ |
| C3 | X | √ | X | √ | X | X | √ | √ | X | √ |
| C4 | X | √ | X | √ | X | X | X | X | X | P |
| C5 | X | √ | X | √ | X | X | √ | √ | X | √ |
| C6 | X | √ | X | √ | X | X | √ | √ | X | √ |
| C7 | X | √ | X | √ | X | X | √ | √ | X | √ |
| C8 | √ | √ | X | √ | X | X | X | √ | X | √ |
| D1 | X | √ | X | √ | X | X | X | X | X | √ |
| D2 | X | √ | X | √ | X | X | √ | X | X | √ |
| D3 | X | √ | X | √ | X | X | √ | X | X | √ |
| D4 | X | X | X | √ | X | X | X | X | X | √ |
| D5 | X | X | X | √ | X | X | X | X | X | √ |
| D6 | X | X | X | √ | X | X | X | X | X | √ |
| D7 | X | X | X | √ | X | √ | X | X | X | √ |
| D8 | X | √ | X | √ | X | X | X | X | X | √ |
| SLC-011 | √ | X | X | X | X | X | X | X | X | √ |
| Sitamaquine | X | √ | X | √ | X | X | √ | √ | X | √ |
√: Active, X: Inactive
Conclusion
In summary, a molecular hybridization technique was used to create the quinoline and sulfonamide molecular hybrids. The proposed derivatives, particularly B5, B3, C5, C3, A5, and D2, which demonstrate strong multi-target fitness across CAIX, AURKA, and AURKB, have intriguing potential as anti-cancer drugs, according to molecular docking studies. In addition, A2, A6, B6, D3, and D6 showed selective inhibition of AURKA and AURKB. Most derivatives comply with Lipinski’s rule, indicating good oral bioavailability, with only a few requiring formulation improvements. Compounds A2, B4, B5, B6, D4, D5, and D6 stand out due to their lower toxicity compared to Sitamaquine, making them safer candidates despite the need to address respiratory toxicity. In conclusion, B5 is the most promising compound among all the derivatives acting as a multi-target anti-cancer agent. Further modification, particularly in addressing respiratory toxicity and improving the GI absorption of certain derivatives, will enhance their suitability for clinical research.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Saini A, Kumar M, Bhatt S, Saini V, Malik A. Cancer causes and treatments. J. Pharm. Sci. Res. 2020, 7,3121-34.
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015, 136, E359-E386.
CrossRef - Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Can J Clin 2018, 68,394-24.
CrossRef - Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, Bray F. Cancer statistics for the year 2020: An overview. International journal of cancer. 2021,149(4),778-89.
CrossRef - Ibrahim DA, Abou El Ella DA, El-Motwally AM, Aly RM. Molecular design and synthesis of certain new quinoline derivatives having potential anticancer activity. European Journal of Medicinal Chemistry. 2015,102,115-31.
CrossRef - Alblewi FF, Alsehli MH, Hritani ZM, Eskandrani A, Alsaedi WH, Alawad MO, Elhenawy AA, Ahmed HY, El-Gaby MS, Afifi TH, Okasha RM. Synthesis and Characterization of a New Class of Chromene-Azo Sulfonamide Hybrids as Promising Anticancer Candidates with the Exploration of Their EGFR, h CAII, and MMP-2 Inhibitors Based on Molecular Docking Assays. International Journal of Molecular Sciences. 2023, 24,16716.
CrossRef - Kardile RA, Sarkate AP, Lokwani DK, Tiwari SV, Azad R, Thopate SR. Design, synthesis, and biological evaluation of novel quinoline derivatives as small molecule mutant EGFR inhibitors targeting resistance in NSCLC: In vitro screening and ADME predictions. European Journal of Medicinal Chemistry. 2023, 245,114889.
CrossRef - M. Antypenko, S.I. Kovalenko, O.V. Karpenko, A.M. Katsev, V.P. Novikov, N. S. Fedyunina, 1-R-2-(1,2,4 triazolo 1,5-c quinazoline-2-ylthio) etanon (ol) s: synthesis, bioluminescence inhibition, molecular docking studies, antibacterial and antifungal activities, Curr. Comput. Aided Drug Des. 2016,12, 29–41.
CrossRef - Yulya, A. Oleksii, N. Inna, B. Galina, K. Sergii, Directed search of anti- inflammatory agents among (3H-Quinazoline-4-ylidene) hydrazides of N-protected amino acids and their heterocyclization products, Antiinflamm. Antiallergy Agents. Med. Chem. 2020,19, 61–73.
CrossRef - Musiol, M. Serda, S.H. Bielowka, J. Polanski, Quinoline-based antifungals, Curr. Med. 2020,17,1960–1973
CrossRef - Neelarapu, J.R. Maignan, C.L. Lichorowic, A. Monastyrskyi, T.S. Mutka, A.N. L. Crue, L.D. Blake, D. Casandra, S. Mashkouri, J.N. Burrows, P.A. Willis, D.E. Kyle, R. Manetsch, Design and synthesis of orally bioavailable piperazine substituted 4 (1H)-Quinolones with potent antimalarial activity: structure-activity and structure- property relationship studies, J. Med. Chem., 2018, 61, 1450–1473,
CrossRef - S.A.A. Salem, G.H. Hegazy, K.E.H. El-Taher, S.M. El-Messery, A.M. Al-Obaid, H. I. El-Subbagh, Synthesis, anticonvulsant activity and molecular modelling study of some new hydrazinecarbothioamide, benzenesulfonohydrazide, and phenacylacetohydrazide analogues of 4(3H)-quinazolinone, Bioorg. Med. Chem. Lett. 2015, 25, 1490–1499
CrossRef - Othman DI, Selim KB, Magda AA, Tantawy AS, Amen Y, Shimizu K, Okauchi T, Kitamura M. Design, synthesis and anticancer evaluation of new substituted thiophene-quinoline derivatives. Bioorganic & Medicinal Chemistry. 2019, 27, 115026.
CrossRef - Selim MR, Zahran MA, Belal A, Abusaif MS, Shedid SA, Mehany A, Elhagali GA, Ammar YA. Hybridized quinoline derivatives as anticancer agents: design, synthesis, biological evaluation, and molecular docking. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 2019, 19, 439-52.
CrossRef - A. Pradeep, R.K. Nagiri, S. Krishana, N.S. Reddy, S. Kanugala, C.G. Kumar, N. J. Babu, T. Ganapathi, N. Banda, Design and synthesis of novel pyrimidine/ hexahydroquinazoline-fused pyrazolo 3,4-b pyridine derivatives. Their biological evaluation and docking studies, Chem. Select. 2019,4, 138–144,
CrossRef - Martorana, G.L. Monica, A. Lauria, Quinoline-based molecules targeting c-met, EGF, and VEGF receptors and the proteins involved in related carcinogenic pathways, Molecules. 2020,25, 4279.
CrossRef - A. Shaheen, A.A. El-Emam, N.S. El-Gohary, Design, synthesis and biological evaluation of new series of hexahydroquinoline and fused quinoline derivatives as potent inhibitors of wild-type EGFR and mutant EGFR (L858R and T790M), Bioorg. Chem. 2020, 105, 104274,
CrossRef - Omidkhah N, Hadizadeh F, Abnous K, Ghodsi R. Synthesis, structure activity relationship and biological evaluation of a novel series of quinoline–based benzamide derivatives as anticancer agents and histone deacetylase (HDAC) inhibitors. Journal of Molecular Structure. 2022, 1267, 133599.
CrossRef - F.A. Mohamed, G.D.A. Abuo-Rahma, Molecular targets and anticancer activity of quinoline–chalcone hybrids: literature review, RSC Adv. 2020, 10, 31139,
CrossRef - Erguc, M.D. Altintop, O. Atli, B. Sever, G. Iscan, G. Gormus, A. Ozdemir, Synthesis and biological evaluation of new quinoline-based thiazolyl hydrazone derivatives as potent antifungal and anticancer agents, Lett. Drug Des. Discov. 2018, 15, 193–202,
CrossRef - Panduranga P, Makam P, Kumar Katari N, Gundla R, Babu Jonnalagadda S, Kumar Tripuramallu B. Molecular Hybrids of Quinoline and Sulfonamide: Design, Synthesis and in Vitro Anticancer Studies. 2024, 14, e202400334.
CrossRef - Khair-ul-Bariyah S, Sarfraz M, Arshad M, Waseem A, Khan HU, Khan S, Sharif A, Farooqi ZH, Ahmed E. Synthesis of 2-aminothiazole sulfonamides as potent biological agents: Synthesis, structural investigations and docking studies. 2024, 10(15).
CrossRef - Wan S, Wu N, Yan Y, Yang Y, Tian G, An L, Bao X. Design, synthesis, crystal structure, and in vitro antibacterial activities of sulfonamide derivatives bearing the 4-aminoquinazoline moiety. Molecular Diversity. 2023, 27, 1243-54.
CrossRef - Zhang SG, Wan YQ, Wen Y, Zhang WH. Novel coumarin 7-carboxamide/sulfonamide derivatives as potential fungicidal agents: design, synthesis, and biological evaluation. 2022, 27, 6904.
CrossRef - Al-Wahaibi LH, Elshamsy AM, Ali TF, Youssif BG, Bräse S, Abdel-Aziz M, El-Koussi NA. Design and synthesis of new dihydropyrimidine/sulphonamide hybrids as promising anti-inflammatory agents via dual mPGES-1/5-LOX inhibition. Frontiers in Chemistry. 2024, 12, 1387923.
CrossRef - Lee CY, Choi H, Park EY, Nguyen TT, Maeng HJ, Mee Lee K, Jun HS, Shin D. Synthesis and anti‐diabetic activity of novel biphenylsulfonamides as glucagon receptor antagonists. Chemical Biology & Drug Design. 2021, 98, 733-50.
CrossRef - Fuentes-Martín R, Ayuda-Durán P, Hanes R, Gallego-Yerga L, Wolterinck L, Enserink JM, Álvarez R, Peláez R. Promising anti-proliferative indolic benzenesulfonamides alter mechanisms with sulfonamide nitrogen substituents. European Journal of Medicinal Chemistry. 2024, 275,116617.
CrossRef - Alblewi FF, Alsehli MH, Hritani ZM, Eskandrani A, Alsaedi WH, Alawad MO, Elhenawy AA, Ahmed HY, El-Gaby MS, Afifi TH, Okasha RM. Synthesis and Characterization of a New Class of Chromene-Azo Sulfonamide Hybrids as Promising Anticancer Candidates with the Exploration of Their EGFR, h CAII, and MMP-2 Inhibitors Based on Molecular Docking Assays. International Journal of Molecular Sciences. 2023, 24(23),16716.
CrossRef - Bukowski K, Marciniak B, Kciuk M, Mujwar S, Mojzych M, Kontek R. Pyrazolo [4, 3-e] tetrazolo [1, 5-b][1, 2, 4] triazine sulfonamides as novel potential anticancer agents: Apoptosis, oxidative stress, and cell cycle analysis. International Journal of Molecular Sciences. 2023, 24(10),8504.
CrossRef - Bourzikat O, El Abbouchi A, Ghammaz H, El Brahmi N, El Fahime E, Paris A, Daniellou R, Suzenet F, Guillaumet G, El Kazzouli S. Synthesis, anticancer activities and molecular docking studies of a novel class of 2-phenyl-5, 6, 7, 8-tetrahydroimidazo [1, 2-b] pyridazine derivatives bearing sulfonamides. 2022, 27(16), 5238.
CrossRef - Shafique I, Saeed A, Ahmed A, Shabir G, Ul-Hamıd A, Khan A, Tüzün B, Kirici M, Taslimi P, Latif M. Exploring the multi-target enzyme inhibition potential of new sulfonamido-thiazoline derivatives; synthesis and computational studies. Results in Chemistry. 2022, 4,100656.
CrossRef - Marciniec K, Nowakowska J, Chrobak E, Bębenek E, Latocha M. Synthesis, Docking, and Machine Learning Studies of Some Novel Quinoline sulfonamides–Triazole Hybrids with Anticancer Activity. 2024, 29(13), 3158.
CrossRef - Panduranga P, Makam P, Kumar Katari N, Gundla R, Babu Jonnalagadda S, Kumar Tripuramallu B. Molecular Hybrids of Quinoline and Sulfonamide: Design, Synthesis and in Vitro Anticancer Studies. 2024, 14, e202400334.
CrossRef - Zieba A, Pindjakova D, Latocha M, Plonka-Czerw J, Kusmierz D, Cizek A, Jampilek J. Design, synthesis, and Anticancer and Antibacterial activities of Quinoline-5-Sulfonamides. 2024, 29(17), 4044.
CrossRef - Kardile RA, Sarkate AP, Lokwani DK, Tiwari SV, Azad R, Thopate SR. Design, synthesis, and biological evaluation of novel quinoline derivatives as small molecule mutant EGFR inhibitors targeting resistance in NSCLC: In vitro screening and ADME predictions. European Journal of Medicinal Chemistry. 2023, 245, 114889.
CrossRef - El-Malah A, Taher ES, Angeli A, Elbaramawi SS, Mahmoud Z, Moustafa N, Supuran CT, Ibrahim TS. Schiff bases as linker in the development of quinoline-sulfonamide hybrids as selective cancer-associated carbonic anhydrase isoforms IX/XII inhibitors: A new regioisomerism tactic. Bioorganic Chemistry. 2023, 131, 106309.
CrossRef - Ma J, Gong GH. Discovery of Novel 3, 4-Dihydro-2 (1 H)-Quinolinone Sulfonamide Derivatives as New Tubulin Polymerization Inhibitors with Anti-Cancer Activity. 2022, 27(5),1537.
CrossRef - Cakmak EB, Kurt BZ, Civelek DO, Angeli A, Akdemir A, Sonmez F, Supuran CT, Kucukislamoglu M. Quinoline-sulfamoyl carbamates/sulfamide derivatives: Synthesis, cytotoxicity, carbonic anhydrase activity, and molecular modelling studies. Bioorganic Chemistry. 2021, 110, 104778.
CrossRef - Shaldam M, Nocentini A, Elsayed ZM, Ibrahim TM, Salem R, El-Domany RA, Capasso C, Supuran CT, Eldehna WM. Development of novel quinoline-based sulfonamides as selective cancer-associated carbonic anhydrase isoform IX inhibitors. International Journal of Molecular Sciences. 2021, 22(20), 11119.
CrossRef - Al-Sanea MM, Elkamhawy A, Paik S, Lee K, El Kerdawy AM, Abbas BS, Roh EJ, Eldehna WM, Elshemy HA, Bakr RB, Farahat IA. Sulfonamide-based 4-anilinoquinoline derivatives as novel dual Aurora kinase (AURKA/B) inhibitors: Synthesis, biological evaluation and in silico insights. Bioorganic & medicinal chemistry. 2020, 28(13),115525.
CrossRef - Koyuncu I, Temiz E, Güler EM, Durgun M, Yuksekdag O, Giovannuzzi S, Supuran CT. Effective Anticancer Potential of a New Sulfonamide as a Carbonic Anhydrase IX Inhibitor Against Aggressive Tumors. Chem Med Chem. 2024, 19, e202300680.
CrossRef - Fadaly WA, Mohamed FE, Nemr MT, Sayed AM, Khalil RG, Zidan TH. Novel benzenesulfonamide derivatives as potential selective carbonic anhydrase IX, XII inhibitors with anti-proliferative activity: Design, synthesis, and in silico studies. Bioorganic Chemistry. 2024, 153,107881.
CrossRef - Mboge MY, McKenna R, Frost SC. Advances in anti-cancer drug development targeting carbonic anhydrase IX and XII. Topics in Anti-Cancer Research. 2016, 5, 3-42.
CrossRef - Pastorekova S, Gillies RJ. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer and Metastasis Reviews. 2019, 15, 65-77.
CrossRef - Kumar S, Rulhania S, Jaswal S, Monga V. Recent advances in the medicinal chemistry of carbonic anhydrase inhibitors. European Journal of Medicinal Chemistry. 2021, 209, 112923.
CrossRef - Chafe, S.C.; Vizeacoumar, F.S.; Venkateswaran, G.; Nemirovsky, O.; Awrey, S.; Brown, W.S.; McDonald, P.C.; Carta, F.; Metcalfe, A.; Karasinska, J.M.; et al. Genome-wide synthetic lethal screen unveils novel CAIX-NFS1/xCT axis as a targetable vulnerability in hypoxic solid tumors. Adv. 2021, 7, eabj0364
CrossRef - T. Supuran, Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors, Expert Opin. Investig. Drugs. 2018, 27 (12), 963–970.
CrossRef - Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting mitosis in cancer: emerging strategies. Mol Cell. 2015, 60, 524–536.
CrossRef - Borisa AC, Bhatt HG. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur J Med Chem. 2017,140,1–19.
CrossRef - Damodaran AP, Vaufrey L, Gavard O, Prigent C. Aurora A kinase is a priority pharmaceutical target for the treatment of cancers. Trends Pharmacol Sci. 2017, 38, 687–700.
CrossRef - Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J. Aurora kinases: novel therapy targets in cancers. 2017, 8, 23937–23954.
CrossRef - Willems E, Dedobbeleer M, Digregorio M, Lombard A, Lumapat PN, Rogister B. The functional diversity of Aurora kinases: a comprehensive review. Cell Div. 2018, 13,7.
CrossRef - Pradhan T, Gupta O, Singh G, Monga V. Aurora kinase inhibitors as potential anticancer agents: Recent advances. European Journal of Medicinal Chemistry. 2021, 221, 113495.
CrossRef - Guan YF, Liu XJ, Yuan XY, Liu WB, Li YR, Yu GX, Tian XY, Zhang YB, Song J, Li W, Zhang SY. Design, synthesis, and anticancer activity studies of novel quinoline-chalcone derivatives. Molecules. 2021, 26(16), 4899.
CrossRef - Ahsan MJ, Gautam K, Ali A, Ali A, Altamimi AS, Salahuddin, Alossaimi MA, Lakshmi SV, Ahsan MF. Synthesis, Anticancer Activity, and In Silico Studies of 5-(3-Bromophenyl)-N-aryl-4 H-1, 2, 4-triazol-3-amine Analogs. Molecules. 2023, 28(19), 6936.
CrossRef
Accepted on: 24 Sep 2025
Second Review by: Dr. Shaheen Begum
Final Approval by: Dr. Tanay Pramanik








![]()




{kind=link}