Prediction of New ESIPT Molecules by Machine Learning and Quantum Modelling
 , Pale Wang-Yang2, Yapara Kanabet 3 and Stève-Jonathan Koyambo-Konzapa4
, Pale Wang-Yang2, Yapara Kanabet 3 and Stève-Jonathan Koyambo-Konzapa41Centre National de Recherche pour le Développement (CNRD), N’Djaména, Chad
2Département de Mathématiques et Physiques, Université Saint Charles Lwanga de Sarh, Sarh, Chad
3Faculté de Sciences Exactes et Appliquées, Université de Moundou, Moundou, Tchad
4Laboratoire Matière, Énergie et Rayonnement (LAMER), Université de Bangui, Bangui, République Centrafricaine
Corresponding Author E-mail: abdallahbrahim88@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/420110
Download this article as:
![]()
The discovery of novel compounds exhibiting Excited State Intramolecular Proton Transfer (ESIPT) is critical for advancing technologies in optoelectronics, biosensors, and smart materials. Conventional approaches dependent on exhaustive quantum chemical simulations are inefficient for exploring expansive molecular libraries. We introduce an integrated computational strategy that combines Density Functional Theory (DFT) with supervised machine learning to efficiently classify ESIPT propensity. Using a curated set of 100 organic molecules, we implemented and compared three classifiers: Random Forest (RF), Support Vector Machine (SVM), and a Deep Neural Network (DNN). The RF model delivered the most robust performance, attaining 94.2% accuracy and a 91.1% F1-score under stratified cross validation. Analysis of feature importance identified key electronic descriptors, including absorption wavelength and LUMO energy, as primary predictors. This hybrid DFT-ML pipeline provides a rapid and reliable method for the virtual screening and targeted design of new ESIPT active materials.
KEYWORDS:Deep Neural Network; DFT/TDDFT; Excited State Intramolecular Proton Transfer; Fluorescence Spectroscopy; Machine Learning; Random Forest; Support Vector Machine
Introduction
The unique photophysical signature of Excited State Intramolecular Proton Transfer (ESIPT) characterized by a large Stokes shift and often dual fluorescence makes it a cornerstone mechanism for designing advanced fluorescent probes and optoelectronic materials.1,2 However, the rational design of new ESIPT capable molecules is bottlenecked by the computational intensity of probing excited state potential energy surfaces with quantum chemical methods like TD-DFT3,4 The ESIPT mechanism involves an ultrafast redistribution of charge following photoexcitation, which dramatically increases the acidity of a proton donor and the basicity of a nearby acceptor, facilitating an intramolecular proton relay.1 This process, depicted in Figure 1, is governed by the energy barrier for proton transfer and the relative stability of the excited normal (N) and tautomeric (T) forms.
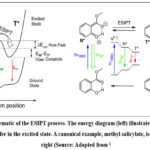 |
Figure 1: Schematic of the ESIPT process. The energy diagram (left) illustrates the favorable proton transfer in the excited state. A canonical example, methyl salicylate, is shown on the right (Source: Adapted from1. |
While the utility of ESIPT is well established, its prediction for new molecular scaffolds remains a formidable challenge. High throughput discovery is hindered by the computational cost of reliably modeling excited-state dynamics. Quantum chemical methods, particularly time dependent density functional theory (TD/DFT), are the standard for probing ESIPT mechanisms but become prohibitively expensive for screening large chemical spaces.3,4 Furthermore, the predictive accuracy of these methods can vary significantly with the choice of exchange correlation functional and the chemical environment.5
This computational difficulty presents a clear opportunity for data driven approaches. Machine learning (ML) has transformed materials discovery by learning complex structure property relationships from existing data, enabling rapid property prediction.6. The successful application of ML to ESIPT prediction hinges on identifying a concise yet informative set of molecular descriptors that can be computed efficiently. We propose that key geometric and electronic features derived from a single, ground state DFT calculation can serve as effective proxies for predicting the complex, excited state ESIPT outcome.
In this study, we establish and validate a practical ML workflow for the binary classification of ESIPT activity. We assembled a balanced dataset of 100 molecules with known ESIPT behavior, computed a tailored suite of DFT based descriptors, and trained three distinct ML models, Random Forest (RF), Support Vector Machine (SVM), and a Deep Neural Network (DNN). Our results not only demonstrate the high predictive accuracy of the RF model but also, through interpretability analysis, reveal the quantum chemical features most indicative of ESIPT propensity, thereby providing actionable guidelines for synthesizing new ESIPT active chromophores.
Methodology
This study aimed to develop a machine learning (ML) framework for the binary classification of ESIPT activity. The overall workflow (Figure 2) integrated initial quantum chemical calculations for descriptor generation with subsequent supervised ML model training and validation.
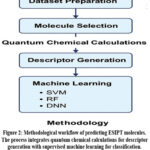 |
Figure 2: Methodological workflow of predicting ESIPT molecules. Click here to View Figure |
This methodology integrates quantum chemical descriptors obtained from Density Functional Theory calculations with supervised machine learning algorithms for the classification tasks.
Molecule Selection and Data Generation
A selection of 100 candidate molecules was based on an in depth review of existing literature and recognized chemical databases. The focus was on organic compounds known for their ability (or lack thereof) to undergo Excited State Intramolecular Proton Transfer. The selected chemical families for investigation include flavones, chromones, oxazoles, and other functionalized aromatic structures, which are likely to exhibit this phenomenon.6,7
All electronic structure calculations were performed to generate molecular descriptors. Ground-state geometry optimizations and subsequent single point energy calculations were conducted using Density Functional Theory (DFT) with the B3LYP hybrid functional and the 6-31G(d,p) basis set, as implemented in the Gaussian 16 software suite.8,9
Spectroscopic properties were also calculated, including electronic transitions and absorption, emission wavelengths, which are essential for understanding the ESIPT process in the studied molecules.10 This set of computational data served as the foundation for the subsequent machine learning analysis.
For each molecule, molecular descriptors were extracted using a combination of geometrical and electronic descriptors. These included distances between atoms involved in proton transfer, bond angles and flatness indices, which are important for activating the ESIPT process 11, as well as ionization energies, electroaffinities, partial charges of atoms, and electron densities in both ground and excited states.12,13 Additionally, excitation energies, absorption wavelengths, and fluorescence energies were calculated, which are essential for determining the suitability of the molecule for optoelectronic applications.14
Each molecule was assigned a definitive label (1) for ESIPT active, (0) for inactive based on consensus from prior experimental or high-level theoretical studies. Molecular descriptors were calculated using DFT methods and included features such as HOMO and LUMO energy levels, dipole moment, bond length and OH group angle, absorption and emission wavelengths, atomic charges (O, H, N), energy gap, and polarizability. Descriptors like the O-H bond length and atomic charges were calculated to capture precisely these photo-induced changes.
The same descriptors were calculated for a set of unlabelled molecules with the aim to predict the ESIPT potential.
Data preparation
Before implementing the machine learning algorithms, the molecular dataset underwent rigorous preprocessing to ensure data quality and consistency. Missing or incomplete entries were identified and removed to avoid biases and errors during model learning. Numerical characteristics such as bond lengths, angles, energies, and loads were standardised to ensure their comparability, thereby enhancing the convergence and performance of the learning algorithms.
Categorical or binary labels showing the presence (1) or absence (0) of an ESIPT behaviour were appropriately encoded for the supervised classification tasks. Feature selection techniques and correlation analyses were conducted to reduce redundancy and focus on the most relevant molecular descriptors. Subsequently, the cleaned and encoded dataset was separated into training and test subsets, ensuring balanced class distributions for reliable model evaluation. To circumvent the issue of overlearning and enhance the model’s generalisation capability, a dimension reduction technique, such as Principal Component Analysis (PCA) was employed.15
Model construction
Three distinct supervised learning algorithms were implemented and compared using scikit-learn and TensorFlow/Keras in Python.
Random Forest algorithm is robust and capable for modelling complex relationships between molecular descriptors and system properties, while exhibiting reduced sensitivity to overlearning.
A random forest is a classification system consists of multiple tree structured classifiers {h(x, θk), k = 1, …}, where the {θk} are independent and identically distributed random vectors and each tree casts a unit vote for the most popular class at input x.16
We used Support Vector Machines with a radial basis function (RBF) kernel to classify molecules based on to their propensity to undergo intramolecular proton transfer. In SVM case, a classification model is built from training samples of the form <xi, yi> where xi = (xi1, xi2, …xim) represents the molecular descriptor vector and yi Є {-1, +1} denotes the binary class label. The algorithm aims to identify an optimal hyperplane that maximises the margin between the two classes. The SVM are particularly well-suited for binary classification problems and perform well in high dimensional descriptor spaces with complex, non linear boundaries17
Deep Neural Networks (DNN) were also investigated to capture deeper, non linear correlations between descriptors and ESIPT activity. DNN model consisted of multiple fully connected layers with RELU activations and dropout for regularisation to mitigate overfitting. DNNs have demonstrated a strong ability to capture abstract feature relationships, especially when handling large and heterogeneous chemical datasets.18
All models were trained and validated using stratified k fold cross validation. Hyperparameters for each algorithm were optimised using grid search to ensure the best performance in terms of classification, accuracy and generalisation capability.
The performance of the models was evaluated using conventional metrics such as precision, recall, F-measure and accuracy. Cross validation was performed to ensure the reliability of the results.19
Subsequent to the training of the machine learning algorithms, were used to predict ESIPT behaviour of new molecules that were not present in the training database. Candidate molecules with optimal performance based on geometric and electronic descriptors were selected for further analysis and experimental testing.20
The results obtained from the modelling were used to classify the new molecules according to their potential to undergo an ESIPT process. This classification was based on predicted values for excitation energies, absorption wavelengths, and tautomer stability.21
Finally, ab initio calculations were carried out to optimise the properties of the molecules identified as having a high ESIPT potential and others. The simulations were used to modify the functional groups and explore structural modifications that could enhance the efficiency of the intramolecular proton transfer process, with potential applications in optoelectronics, sensors, and photonic devices.22,23
Results And Discussions
Machine learning algorithms were trained and evaluated on the curated dataset of 100 molecules.
Predictive Model Performance
Classification models were evaluated using several measures, including precision, F1 score, precision, recall and area under the curve (AUC).
The performance metrics summarized in Table 1 reveal that all three ML classifiers successfully learn the structure-property relationships governing ESIPT from our DFT derived descriptor set. The Random Forest (RF) model emerged as the optimal classifier for this task, achieving a 94.2% accuracy and a 91.1% F1-score. Its superior performance over the Support Vector Machine (SVM) and Deep Neural Network (DNN) can be attributed to its inherent robustness against overfitting on our curated dataset of 100 molecules and its ability to handle non-linear relationships without extensive hyperparameter tuning. The marginally lower recall of the SVM model suggests a more conservative classification bias, potentially minimizing false positives at the expense of missing some true ESIPT candidates a trade off that may be desirable for high confidence virtual screening.
Table 1: Performance metrics of the machine learning models
| Models | Precision (%) | Recall (%) | F-score (%) | Accuracy (%) |
| Random Forests | 92.5 | 89.8 | 91.1 | 94.2 |
| SVM | 90.3 | 87.6 | 88.9 | 93.5 |
| Deep Neural Network | 79.4 | 83.2 | 85.3 | 90.1 |
Confusion Matrix Analysis
The normalized confusion matrix for the optimal Random Forest model (Figure 2) reveals a high proportion of correct predictions along the main diagonal, which aligns with the strong performance metrics reported in Table 1. Specifically, the matrix demonstrates a high true positive rate, indicating the model’s effectiveness in correctly identifying ESIPT-active molecules a key requirement for a discovery tool. The corresponding low false positive rate is particularly notable, as it minimizes the risk of erroneously allocating experimental resources to synthesize inactive candidates. The minimal number of false negatives suggests that the model rarely misses genuinely ESIPT active structures, ensuring comprehensive coverage in virtual screening campaigns. This performance profile confirms the model’s reliability and suitability for prioritizing candidates for further investigation.
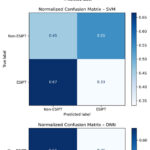 |
Figure 3: Normalized confusion matrix showing the model’s classification performance for ESIPT prediction. Click here to View Figure |
Support Vector Machines (SVMs) showed effectiveness in handling borderline cases, while Deep Neural Networks (DNN) proved to be superior in capturing complex non-linear relationships within the data, albeit requiring a longer training period.
Model Discriminatory
ROC curves were generated for each classifier to assess discrimination between ESIPT and non-ESIPT molecules across thresholds. The RF’s AUC, significantly above 0.9, confirms its excellent ability to distinguish between the two classes across all classification thresholds.
 |
Figure 4: Receiver Operating Characteristic (ROC) curves. Click here to View Figure |
Feature importances analysis
Feature importance analysis allows us to identify the molecular descriptors that significantly contribute to the model’s decision making process in predicting ESIPT activity. In our study, the Random Forest classifier revealed that features related to electronic properties such as Absorption wavelength, LUMO energy, Emission wavelength, excitation energy, and atomic charges had the highest importance scores.
Geometric descriptors also played a crucial role, particularly the distances between O-H and H-N bonds, as well as the degree of dipole moment. Spectroscopic features contributed moderately to the predictions.
The feature importance analysis from the leading RF model (Figure 4) provides critical chemical insight beyond predictive accuracy. The high ranking of the S₁-state LUMO energy and absorption wavelength underscores the pivotal role of excited state charge redistribution and energy stabilization in facilitating proton transfer. Crucially, the prominence of the change in natural charge on the proton donor atom upon photoexcitation quantitatively validates the long held qualitative principle of photoacidity driving ESIPT. Furthermore, the significant contribution of ground-state geometric descriptors, such as the O-H…N bond distance, confirms that the ML model correctly identifies the pre-organized hydrogen-bonded scaffold as a necessary precondition. This interpretable output transitions our workflow from a black box predictor to a tool for hypothesis generation, offering concrete guidelines, target structures should feature a strong intramolecular hydrogen bond and substituents that stabilize the LUMO and enhance charge transfer upon excitation.
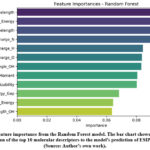 |
Figure 5: Feature importance from the Random Forest model. Click here to View Figure |
PCA Distribution and Class Separation
To visualize the structure of the descriptor space and evaluate the extent to which ESIPT and non-ESIPT molecules form separable groups, principal component analysis (PCA) was applied as a dimensionality reduction step. By projecting the high dimensional descriptor data onto the first two principal components, which capture the majority of the variance in the dataset, we obtained a two-dimensional representation that facilitates pattern recognition and class discrimination.
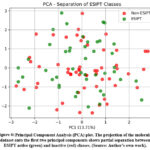 |
Figure 6: Principal Component Analysis (PCA) plot. Click here to View Figure |
The PCA analysis depicted in Figure 5 illustrates a clear grouping of ESIPT and non-ESIPT molecules. This distinct separation indicates that the chosen descriptors effectively capture the chemical differences relevant to proton transfer behavior. While some overlap between the two classes is present, the observed separation validates the molecular features used and supports their application in machine learning classification models.
In addition, PCA can mitigate issues such as multicollinearity and descriptor noise, which can improve the robustness and generalizability of downstream predictive models. From a modelling perspective, focusing on dominant sources of variation can also reduce overfitting risk and improve interpretability.
Prediction on New Unlabeled Structures
The validated Random Forest model was deployed as a screening tool to predict the ESIPT potential of 50 novel, unlabeled molecular structures. These candidate molecules were represented using the same suite of 22 DFT derived descriptors as the training set. The classifier provides a binary prediction alongside a calibrated probability score, enabling the prioritization of candidates with high predicted confidence (>0.9) for ESIPT activity. This rapid in silico screening allows for the efficient evaluation of large virtual libraries, focusing subsequent, more computationally intensive TD-DFT validation and experimental synthesis efforts on the most promising leads, thereby dramatically accelerating the discovery pipeline compared to exhaustive quantum chemical screening alone.
Conclusion
In conclusion, we have established an integrated DFT/ML pipeline for the efficient and interpretable prediction of ESIPT activity. Using a curated dataset of 100 molecules from established ESIPT families, we demonstrate that a Random Forest classifier, trained on a tailored set of 22 quantum chemical descriptors, achieves high classification performance (94.2% accuracy, 91.1% F1-score). Beyond its utility as a virtual screening tool, the model’s interpretability delivers key chemical insights: the critical importance of the S1 state LUMO energy and the photoinduced change in donor atom charge quantitatively underscores the role of excited state charge redistribution in driving proton transfer. Furthermore, the model correctly identifies a pre organized hydrogen bonded geometry as a fundamental prerequisite. These insights provide concrete, quantum mechanically informed guidelines for the targeted design of new ESIPT chromophores. The primary limitation of this proof of concept study is the dataset size; future work will expand chemical diversity and explore advanced molecular representations like graph neural networks. This workflow establishes a practical paradigm for accelerating the discovery of next generation ESIPT materials for sensing and optoelectronics.
Acknowledgement
The authors acknowledge the Centre National de Recherche pour le Développement (CNRD), N’Djaména, for providing computational resources and infrastructure.
Funding Sources
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data Availability Statement
The dataset of molecular structures, calculated descriptors, and labels generated and analyzed during this study is available from the corresponding author upon reasonable request.
Author’s Contributions
Abdallah Brahim Elhadj Ali : Conceptualization, Methodology, Formal analysis, Writing original draft.
Pale Wang-Yang : Software, Data curation, Validation.
Yapara Kanabet: Software, Data curation, Validation.
Stève-Jonathan Koyambo-Konzapa : Visualization, Writing review & editing.
References
- Hem C. Joshi, Liudmil Antonov, Excited-State Intramolecular Proton Transfer: A Short Introductory Review, Molecules 2021, 26(5), 1475; https://doi.org/10.3390/molecules26051475 .
CrossRef - Tavakol, H., Keshavarzipour, F. A DFT study of inter- and intramolecular proton transfer in 2-selenobarbituric acid tautomers. Struct Chem 26, 1049-1057 (2015). https://doi.org/10.1007/s11224-015-0567-y.
CrossRef - Yue Liu, Tianlu Zhao, Wangwei Ju, Siqi Shi, Materials discovery and design using machine learning, Journal of Materiomics, 3, 3, 2017, 159-177, https://doi.org/10.1016/j.jmat.2017.08.002.
CrossRef - Julia Westermayr, Philipp Marquetand, Machine Learning for Electronically Excited States of Molecules, Chem. Rev. 2021, 121, 9873-9926, DOI: 10.1021/acs.chemrev.0c00749.
CrossRef - Rama Vasudevan, Ghanshyam Pilania; Prasanna V. Balachandran, Machine learning for materials design and discovery, J. Appl. Phys. 129, 070401 (2021), https://doi.org/10.1063/5.0043300.
CrossRef - Kasha, M, Characterization of electronic transitions in complex molecules. Discussions of the Faraday Society, 9, 1950.
CrossRef - Ling Chen, Peng-Yan Fu, Hai-Ping Wang, Mei Pan, Excited-State Intramolecular Proton Transfer (ESIPT) for Optical Sensing in Solid State, Advanced optical materials, https://doi.org/10.1002/adom.202001952.
CrossRef - Becke, A. D., Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993), https://doi.org/10.1063/1.464913.
CrossRef - Thomas W. Keal, David J. Tozer, Semiempirical hybrid functional with improved performance in an extensive chemical assessment, J. Chem. Phys. 123, 121103 (2005), https://doi.org/10.1063/1.2061227.
CrossRef - Jianzhang Zhao, Shaomin Ji, Yinghui Chen, Huimin Guo, Pei Yang, Excited state intramolecular proton transfer (ESIPT): from principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials, Physical Chemistry Chemical Physics, 25, 2012, 8753-9236.
CrossRef - Steve Scheiner, Theoretical Studies of Excited State Proton Transfer in Small Model Systems, J. Phys. Chem. A, 2000, 104, 25, 5898-5909, https://doi.org/10.1021/jp000125q.
CrossRef - Michael Kasha, Energy Transfer, Charge Transfer, and Proton Transfer in Molecular Composite Systems, Physical and Chemical Mechanisms in Molecular Radiation Biology, 58, 231-255.
- Jessica M. J. Swanson, Jack Simons, Role of Charge Transfer in the Structure and Dynamics of the Hydrated Proton, J. Phys. Chem. B, 2009, 113, 15, 5149-5161, https://doi.org/10.1021/jp810652v.
CrossRef - Yajie Zhang, Changjiao Shang, Yunjian Cao, Min Ma, Chaofan Sun, Insights into the photophysical properties of 2-(2’-hydroxyphenyl) benzazoles derivatives: Application of ESIPT mechanism on UV absorbers, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 280, 2022, 121559, https://doi.org/10.1016/j.saa.2022.121559.
CrossRef - Jolliffe, I. T., Principal Component Analysis for Special Types of Data, 338-372, Springer Series in Statistics, Springer, New York.
- Breiman L., Random forests, Machine Learning, 45, 5-32, (2001).
CrossRef - Cortes C., Vapnik, V., Support-vector networks, Machine Learning, 20(3), 273-297, (1995).
CrossRef - LeCun Y., et al., Deep learning, Nature, 521, 436-444(2015).
CrossRef - Kohavi R., A study of cross-validation and bootstrap for accuracy estimation and model selection, International Joint Conference on Artificial Intelligence (IJCAI), 1137-1143(1995).
- Alexandre Varnek, Igor Baskin, Machine Learning Methods for Property Prediction in Chemoinformatics: Quo Vadis?, J. Chem. Inf. Model. 2012, 52, 6, 1413-1437, https://doi.org/10.1021/ci200409x.
CrossRef - Luís F. B. Fontes, João Rocha, Artur M. S. Silva, Samuel Guieu, Excited-State Proton Transfer in Luminescent Dyes: From Theoretical Insight to Experimental Evidence, chemistry a european journal, 29, 57, 2023, https://doi.org/10.1002/chem.202301540.
CrossRef - Jean Nunes Laner, Henrique de Castro Silva Junior, Fabiano Severo Rodembusch and Eduardo Ceretta Moreira, New insights on the ESIPT process based on solid-state data and state-of-the-art computational methods, Physical Chemistry Chemical Physics, 2, 2021.
Accepted on: 17 Sep 2025
Second Review by: Dr. Asif Khan
Final Approval by: Dr. Ioana Stanciu








![]()




{kind=link}