Docking and Theoretical Studies of Benzoxazole Derivative with Enhanced NLO and Anti-Hyperlipidemic Activity
 and Vishal Dubey2*
and Vishal Dubey2*1Department of Pharmaceutical Chemistry, Maharana Pratap College of Pharmaceutical Sciences, Kanpur, Uttar Pradesh, India
2Department of Pharmaceutical Chemistry, Naraina Vidya Peeth Group of Institution, Panki, Kanpur, Uttar Pradesh, India
Corresponding Author E-mail: vishal.9dec@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/420120
Download this article as:
![]()
Antihyperlipidemic activity refers to a compound’s ability to lower lipid levels, particularly cholesterol and triglycerides, thereby reducing the risk of cardiovascular diseases such as atherosclerosis and coronary artery disease. Benzoxazole derivatives are known for their wide-ranging pharmacological and optoelectronic applications. Computational investigations were performed using DFT and TD-DFT (B3LYP/6-311++G(d,p)) to study optimized geometry, FMO, DOS, MEP, and NLO properties in gas and aqueous phases. Molecular docking (AutoDock Vina, PyRx) was employed against the ACAT enzyme (PDB ID: 1DQ9) to assess bioactivity, with interactions visualized in BIOVIA Discovery Studio. FMO and DOS analyses revealed a narrow HOMO–LUMO gap (4.7185 eV in gas, 4.6643 eV in water), indicating enhanced charge transfer and electronic delocalization in polar media. The MEP map identified active electrophilic and nucleophilic regions, aligning with global reactivity descriptors. TD-DFT results showed π→π* transitions with slight red shifts in aqueous phase, confirming solvent-induced stabilization. Notably, NMBA exhibited an exceptional NLO response with a first-order hyperpolarizability (βtot = 20447.47x10-33 e.s.u.), about 61 times greater than urea, highlighting its strong optical activity. Molecular docking against 1DQ9 protein revealed a high binding affinity (-7.50 kcal/mol) via hydrogen bonding, π–alkyl, and hydrophobic interactions, affirming NMBA’s potential as an effective anti-cholesterol agent. Theoretical and docking results highlight NMBA as a promising multifunctional molecule with superior NLO properties and significant antihyperlipidemic potential, bridging optoelectronic and pharmaceutical applications.
KEYWORDS:Benzoxazole; DFT; TD-DFT; DOS; Molecular docking; NLO
Introduction
Antihyperlipidemic activity refers to the ability of a compound to reduce lipid levels, particularly cholesterol and triglycerides, thereby lowering the risk of atherosclerosis, coronary artery disease, and stroke.1,2 Key therapeutic targets include ACAT and HMG-CoA reductase, and recent efforts focus on developing novel synthetic and natural inhibitors to overcome statin intolerance.3 Notably, benzoxazole-based compounds have previously shown promising ACAT inhibitory activity, supporting their potential as antihyperlipidemic agents.4-6
The benzoxazole nucleus, composed of a fused benzene and oxazole ring, is a key heterocyclic framework widely explored in medicinal and materials chemistry.7-10 Its strong π-electron delocalization and planarity confer unique physicochemical and biological properties.11 Benzoxazole derivatives display diverse pharmacological activities i.e. antimicrobial, anticancer, anti-inflammatory, antiviral, antioxidant, antitubercular, and antihyperlipidemic attributed to their ability to form hydrogen bonds, π-π stacking, and hydrophobic interactions with biological targets.12-14
Benzoxazole derivatives continue to represent a privileged scaffold in medicinal chemistry due to their diverse pharmacological activities and structural versatility. Figure 1 illustrates a series of benzoxazole-based compounds (1-5) exhibiting significant biological potential across various therapeutic areas. Compound 1 acts as an anti-HIV agent.15, while compound 2 is identified as an anti-inflammatory agent.16 Compound 3 displays promising anticancer activity through possible apoptotic pathways.17 while compound 4 functions as a selective COX-2 inhibitor.18 Moreover, compound 5 exhibits antibacterial properties.19 The highlighted structure in the dashed box represents a novel benzoxazole analog designed in this work, aimed at enhancing biological potency through rational modification of the core scaffold. Beyond medicinal applications, benzoxazole derivatives hold great significance in materials science due to their superior optoelectronic properties, such as high thermal stability, strong fluorescence, and efficient intramolecular charge transfer.20-22 These features make them attractive candidates for nonlinear optical (NLO) materials, OLEDs, and photovoltaic devices.
 |
Figure 1: Some examples of bioactive benzoxazole derivatives. Click here to View Figure |
However, despite extensive synthetic and biological studies, derivatives like N-ethyl-N-(2-(2-(5-methylbenzo[d]oxazol-2-yl)hydrazineyl)ethyl)aniline (NMBA) remain theoretically underexplored. A lack of computational investigation limits insight into their electronic structure, reactivity, and stability. Modern quantum chemical tools, especially Density Functional Theory (DFT) and Time-Dependent DFT (TD-DFT), provide powerful means to elucidate these molecular characteristics and rationalize their structure-property relationships.23,24
Density Functional Theory (DFT) calculations at the B3LYP/6-311++G(d,p) level were performed to optimize the molecular structure of NMBA and evaluate its electronic and optical properties through FMO, DOS, MEP, and NLO analyses. Solvent effects were modelled using the PCM approach, while TD-DFT calculations provided insights into excited-state transitions and absorption spectra. Molecular docking using PyRx–AutoDock Vina against receptor 1DQ9, validated via Ramachandran analysis, revealed the compound’s strong binding affinity and stability. This integrated DFT–TDDFT–docking study elucidates NMBA’s electronic behaviour and bio interaction potential, emphasizing its dual applicability in optoelectronic and pharmaceutical fields.
Computational Details
Density functional theory (DFT) calculations
All quantum chemical calculations were carried out using the Gaussian 09W software package.25 The molecular geometry of (NMBA) was fully optimized in the gas phase without any symmetry constraints using the B3LYP hybrid functional in conjunction with the 6-311++G(d,p) basis set.26,27 Frequency calculations confirmed that the optimized geometry corresponds to a true minimum on the potential energy surface, as no imaginary frequencies were observed. All visualizations of optimized structures, FMOs, and MEP surfaces were generated using GaussView 6.0 28 and ChemCraft 1.8 29 software.
Molecular docking studies
Molecular docking is a robust computational technique used to predict the most favourable orientation of ligands within protein active sites, providing insight into interaction strength and binding specificity.30 In this study, docking analyses were carried out to explore the antihyperlipidemic potential of the benzoxazole derivative (NMBA) against the ACAT enzyme (PDB ID: 1DQ9.31 a pivotal target in cholesterol metabolism. Inhibition of ACAT and HMG-CoA reductase (HMGR) is known to lower cholesterol esterification and enhance LDL clearance, thereby maintaining lipid homeostasis. Considering the therapeutic limitations of conventional statins, AutoDock Vina (PyRx) was employed to simulate NMBA-ACAT interactions and predict its potential mechanism of action. Protein preparation involved the removal of crystallographic water molecules and the addition of hydrogen atoms to optimize binding site geometry. The obtained docking results provided binding affinities and optimized ligand conformations, which were further analysed using BIOVIA Discovery Studio Visualizer 2025.32 This analysis enabled visualization of key hydrogen bonding, hydrophobic, and π–π stacking interactions, offering detailed insights into the binding stability and interaction mechanism of the NMBA–ACAT complex.
Results and Discussion
Optimized geometry
The optimized molecular geometry of NMBA was obtained at the B3LYP/6-311++G(d,p) level of theory in both gas and aqueous phases. The equilibrium geometrical parameters, including selected bond lengths and bond angles, are summarized in Tables 1 and 2, respectively. The optimized structure (Figure 2) reveals that NMBA maintains a nearly planar conformation around the benzoxazole and hydrazine linkage, whereas the ethyl aniline fragment exhibits slight torsional flexibility due to steric and electronic interactions.
 |
Figure 2: Optimized molecular structure of title compound in gas and water phase. Click here to View Figure |
The optimized geometrical parameters reveal minimal differences between the gas and solvent phases, indicating the structural rigidity of the NMBA framework. The C-C bond lengths within the benzoxazole ring (1.378-1.416 Å) are characteristic of aromatic conjugation, while slight elongation of bonds such as R(3-4), R(4-5), R(5-6), and R(6-7) in the aqueous phase is attributed to solvent-induced polarization and partial π-electron delocalization. The C-N bonds linking the benzoxazole and hydrazine moieties (e.g., R(3-10) = 1.4543 Å in gas, 1.4572 Å in water) are marginally longer than typical C-N single bonds, suggesting partial double-bond character due to conjugation between the benzoxazole nitrogen and the hydrazine lone pair. The N-N linkage within the hydrazine bridge further supports resonance delocalization across the donor-acceptor framework, promoting efficient π-electron flow. Meanwhile, C-H and N-H bonds (e.g., R(1-26), R(1-27), R(2-28)) exhibit negligible changes (<0.01 Å) upon solvation, reflecting minimal solvent influence on peripheral hydrogens. Overall, solvation produces only minor geometric adjustments while enhancing dipolar stabilization through dielectric screening, with slight C-N and C-O bond contraction in water suggesting additional stabilization via hydrogen bonding and dipole–dipole interactions with the solvent continuum.
The calculated bond angles of NMBA exhibit remarkable consistency between gas and solvent phases, confirming its structural stability. Angles within the benzoxazole core (e.g., A(3-4-5) ≈ 121°) align with typical sp² hybridization, supporting its aromatic nature. The N-C-N and C-N-C angles in the hydrazine linkage show slight pyramidalization due to lone-pair interactions and partial conjugation. Minimal deviations in the ethyl aniline unit upon solvation indicate negligible solvent influence on local geometry. Minor angle expansions at donor sites reflect subtle structural relaxation in polar media from enhanced charge delocalization.
Table 1: Bond lengths of title compound.
| Atoms | Gas | Water | Atoms | Gas | Water | Atoms | Gas | Water |
| R(1-2) | 1.5349 | 1.5341 | R(7-33) | 1.0832 | 1.0834 | R(15-16) | 1.3892 | 1.3888 |
| R(1-3) | 1.4614 | 1.4651 | R(8-9) | 1.3909 | 1.3918 | R(16-17) | 1.3974 | 1.3974 |
| R(1-26) | 1.0951 | 1.0944 | R(8-34) | 1.0849 | 1.0851 | R(16-21) | 1.378 | 1.3781 |
| R(1-27) | 1.0926 | 1.0916 | R(9-35) | 1.081 | 1.0809 | R(17-18) | 1.3927 | 1.3936 |
| R(2-28) | 1.0919 | 1.0923 | R(10-11) | 1.5373 | 1.5373 | R(17-23) | 1.3976 | 1.3987 |
| R(2-29) | 1.0927 | 1.0925 | R(10-36) | 1.0959 | 1.0946 | R(18-19) | 1.4003 | 1.4021 |
| R(2-30) | 1.0945 | 1.0941 | R(10-37) | 1.0926 | 1.0918 | R(18-40) | 1.0839 | 1.0843 |
| R(3-4) | 1.3927 | 1.3877 | R(11-12) | 1.4704 | 1.4717 | R(19-20) | 1.4067 | 1.4071 |
| R(3-10) | 1.4543 | 1.4572 | R(11-38) | 1.0917 | 1.0914 | R(19-22) | 1.5119 | 1.512 |
| R(4-5) | 1.4133 | 1.4163 | R(11-39) | 1.0964 | 1.0966 | R(20-21) | 1.3989 | 1.4004 |
| R(4-9) | 1.4131 | 1.4161 | R(12-13) | 1.408 | 1.4074 | R(20-41) | 1.0845 | 1.0844 |
| R(5-6) | 1.3907 | 1.3916 | R(12-25) | 1.0173 | 1.0172 | R(21-42) | 1.0827 | 1.0827 |
| R(5-31) | 1.0808 | 1.0807 | R(13-14) | 1.3663 | 1.3586 | R(22-43) | 1.0955 | 1.0954 |
| R(6-7) | 1.393 | 1.3949 | R(13-24) | 1.01 | 1.0102 | R(22-44) | 1.0926 | 1.0922 |
| R(6-32) | 1.0849 | 1.0851 | R(14-15) | 1.369 | 1.3659 | R(22-45) | 1.0923 | 1.0922 |
| R(7-8) | 1.3928 | 1.3948 | R(14-23) | 1.2955 | 1.3022 |
Table 2: Bond angles of title compound.
| Atoms | Gas | Water | Atoms | Gas | Water | Atoms | Gas | Water |
| A(2-1-3) | 115.0661 | 115.0567 | A(5-6-32) | 118.7026 | 118.6334 | A(14-15-16) | 103.264 | 103.635 |
| A(2-1-26) | 109.857 | 109.9022 | A(7-6-32) | 119.9996 | 120.0082 | A(14-23-17) | 103.718 | 103.796 |
| A(2-1-27) | 109.2463 | 109.2447 | A(6-7-8) | 118.1833 | 118.1038 | A(15-16-17) | 107.309 | 107.294 |
| A(1-2-28) | 111.4568 | 111.6327 | A(6-7-33) | 120.9059 | 120.9482 | A(15-16-21) | 128.776 | 128.746 |
| A(1-2-29) | 111.183 | 111.1476 | A(8-7-33) | 120.908 | 120.9456 | A(17-16-21) | 123.913 | 123.958 |
| A(1-2-30) | 110.0925 | 109.7953 | A(7-8-9) | 121.2687 | 121.3265 | A(16-17-18) | 119.504 | 119.496 |
| A(3-1-26) | 109.2689 | 109.2409 | A(7-8-34) | 120.0004 | 120.0194 | A(16-17-23) | 109.079 | 109.016 |
| A(3-1-27) | 107.1535 | 107.0328 | A(9-8-34) | 118.7294 | 118.653 | A(16-21-20) | 115.793 | 115.801 |
| A(1-3-4) | 121.0442 | 121.1291 | A(8-9-35) | 118.213 | 118.2127 | A(16-21-42) | 122.329 | 122.366 |
| A(1-3-10) | 117.7443 | 117.6007 | A(11-10-36) | 109.7866 | 109.7948 | A(18-17-23) | 131.411 | 131.484 |
| A(26-1-27) | 105.8167 | 105.9396 | A(11-10-37) | 108.5695 | 108.7119 | A(17-18-19) | 118.561 | 118.56 |
| A(28-2-29) | 107.9015 | 108.1342 | A(10-11-12) | 108.693 | 108.7077 | A(17-18-40) | 120.334 | 120.629 |
| A(28-2-30) | 108.0985 | 107.9748 | A(10-11-38) | 109.8647 | 109.8544 | A(19-18-40) | 121.106 | 120.811 |
| A(29-2-30) | 107.9799 | 108.0298 | A(10-11-39) | 110.3499 | 110.3147 | A(18-19-20) | 119.937 | 119.911 |
| A(4-3-10) | 121.2071 | 121.2701 | A(36-10-37) | 106.0031 | 106.18 | A(18-19-22) | 120.119 | 120.075 |
| A(3-4-5) | 121.4571 | 121.4892 | A(12-11-38) | 107.2941 | 107.3888 | A(20-19-22) | 119.931 | 120.004 |
| A(3-4-9) | 121.6615 | 121.7565 | A(12-11-39) | 112.7601 | 112.5538 | A(19-20-21) | 122.291 | 122.274 |
| A(3-10-11) | 114.2788 | 114.0673 | A(11-12-13) | 113.3245 | 113.3225 | A(19-20-41) | 118.974 | 119.013 |
| A(3-10-36) | 109.3929 | 109.5774 | A(11-12-25) | 110.702 | 110.962 | A(19-22-43) | 111.056 | 110.977 |
| A(3-10-37) | 108.4711 | 108.1947 | A(38-11-39) | 107.8111 | 107.9597 | A(19-22-44) | 111.417 | 111.352 |
| A(5-4-9) | 116.8795 | 116.7514 | A(13-12-25) | 109.5794 | 109.4927 | A(19-22-45) | 111.357 | 111.341 |
| A(4-5-6) | 121.1642 | 121.2087 | A(12-13-14) | 122.4429 | 123.1053 | A(21-20-41) | 118.735 | 118.714 |
| A(4-5-31) | 120.5432 | 120.5039 | A(12-13-24) | 114.4486 | 114.9811 | A(20-21-42) | 121.879 | 121.833 |
| A(4-9-8) | 121.1916 | 121.2383 | A(14-13-24) | 114.0398 | 115.3509 | A(43-22-44) | 107.395 | 107.366 |
| A(4-9-35) | 120.5923 | 120.5478 | A(13-14-15) | 116.4463 | 116.667 | A(43-22-45) | 107.419 | 107.38 |
| A(6-5-31) | 118.2926 | 118.2874 | A(13-14-23) | 126.9147 | 127.065 | A(44-22-45) | 108.004 | 108.24 |
| A(5-6-7) | 121.2976 | 121.3581 | A(15-14-23) | 116.6221 | 116.255 |
Frontier molecular orbital (FMO) and global reactivity analysis
The frontier molecular orbitals (HOMO and LUMO) are key indicators of a molecule’s electronic behaviour, stability, and reactivity.33 The HOMO-LUMO energy gap reflects charge-transfer efficiency and chemical responsiveness. Along with, global descriptors such as ionization potential, hardness, and electrophilicity provide quantitative insight into electron-donating and accepting tendencies, collectively revealing the molecule’s reactivity and potential in optoelectronic and biological applications.34,35 The computed EHOMO values are -5.4139 eV (gas) and -5.4447 eV (water), while ELUMO values are -0.6955 eV (gas) and -0.7804 eV (water), showing slight stabilization in the aqueous phase due to solvent polarity (table 3). The energy gap (ΔE) of 4.7185 eV (gas) and 4.6643 eV (water) indicate marginal narrowing upon solvation, suggesting enhanced polarizability, chemical softness, and improved charge-transfer potential features beneficial for NLO and bio-interaction properties (figure 3).
The ionization potential (5.4139 eV in gas, 5.4447 eV in water) indicates moderate electronic stability and a fair electron-donating tendency of NMBA. The electron affinity slightly increases from 0.6955 eV to 0.7804 eV in water, reflecting improved electron-accepting ability in a polar medium. A minor decrease in chemical hardness (2.3592 to 2.3322 eV) and an increase in softness (0.2119 to 0.2144 eV⁻¹) suggest enhanced polarizability and electronic flexibility, favourable for strong NLO and bioactive responses. The rise in electronegativity (3.0547 to 3.1126 eV) and the more negative chemical potential (-3.0547 to -3.1126 eV) further indicate increased electron-attracting capacity. Additionally, the electrophilicity index (1.9777 to 2.0771 eV) and maximum charge transfer index (ΔNmax: 1.2948 to 1.3346) confirm stronger electrophilic and charge-transfer tendencies in aqueous phase, consistent with enhanced reactivity and improved protein-binding potential.
Table 3: Global reactivity descriptors in gas and water phase.
| Parameters | Gas | Water | Units |
| EHOMO | -5.4139 | -5.4447 | eV |
| ELUMO | -0.6955 | -0.7804 | eV |
| Energy band gap (∆E= ELUMO-EHOMO) | 4.7185 | 4.6643 | eV |
| Ionization potential (I = -EHOMO) | 5.4139 | 5.4447 | eV |
| Electron affinity (A = -ELUMO) | 0.6955 | 0.7804 | eV |
| Electronegativity (χ = (I + A)/2) | 3.0547 | 3.1126 | eV |
| Chemical hardness (η = (I-A)/2) | 2.3592 | 2.3322 | eV |
| Chemical softness (δ= 1/2h) | 0.2119 | 0.2144 | eV-1 |
| Chemical potential (μ= -(I+A)/2) = -c | -3.0547 | -3.1126 | eV |
| Electrophilicity index (ω= m2/2h | 1.9777 | 2.0771 | eV |
| Maximum charge transfer index (∆Nmax = -μ/η) | 1.2948 | 1.3346 |
Density of states (DOS) analysis
The Density of States (DOS) analysis (Figure 4), computed at the B3LYP/6-311++G(d,p) level, provides deeper insight into the electronic structure of NMBA in both gas and aqueous phases. The occupied orbitals (green) lie below the Fermi level, while the unoccupied orbitals (red) appear above it. The HOMO is primarily localized on the benzo[d]oxazole and hydrazine regions, indicating strong electron-donating capability, whereas the LUMO is mainly distributed over the aniline moiety, signifying electron-accepting characteristics. The calculated energy gaps of 4.7185 eV (gas) and 4.6643 eV (water) confirm a moderate donor-acceptor interaction and efficient charge transfer, highlighting NMBA’s potential for optoelectronic and bioactive applications.
 |
Figure 3: FMO’s of title compound in gas and water phase. Click here to View Figure |
 |
Figure 4: Density of states (DOS) spectrum in gas and water phase. Click here to View Figure |
Molecular electrostatic potential (MEP) analysis
The Molecular Electrostatic Potential (MEP) map (Figure 5) of title compound, computed at the B3LYP/6-311++G(d,p) level, illustrates the charge distribution across the molecular surface in gas and aqueous medium. The red regions indicate electron-rich sites associated with negative potential, mainly localized around oxygen and nitrogen atoms, suggesting preferred sites for electrophilic attack. Conversely, the blue regions represent electron-deficient zones, favourable for nucleophilic interactions. The distinct variation in electrostatic potential confirms the molecule’s strong intramolecular charge transfer and supports its potential reactivity in biological recognition and interaction processes.
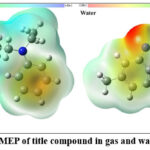 |
Figure 5: MEP of title compound in gas and water phase. Click here to View Figure |
Table 4: The maximum wavelength (λmax), excitation energy, oscillator strength (f), and major contributions in gas and water phase.
| No. | Energy (eV) | λmax (nm) | f | Assignment ≥ 10% |
| Gas | ||||
| 1 | 4.728 | 262.239 | 0.0519 | HOMO->L+8 (61%) |
| 2 | 4.760 | 260.476 | 0.0202 | HOMO->LUMO (71%) |
| 3 | 4.994 | 248.256 | 0.2024 | H-1->LUMO (14%), H-1->L+2 (13%), H-1->L+5 (34%) |
| Water | ||||
| 1 | 4.633 | 267.623 | 0.0659 | HOMO->L+6 (36%), HOMO->L+7 (53%) |
| 2 | 4.846 | 255.822 | 0.0288 | HOMO->LUMO (64%), HOMO->L+2 (16%) |
| 3 | 4.966 | 249.681 | 0.2648 | H-1->L+1 (57%) |
TD-DFT (UV-Vis) analysis
Time-dependent DFT (TD-DFT) analysis at the B3LYP/6-311++G(d,p) level was performed to examine the electronic excitation behaviour of NMBA in both gas and aqueous phases (Figure 6). Three major π→π* transitions were observed at 262, 260, and 248 nm in the gas phase, with the most intense at 248 nm (f = 0.2024). In the aqueous phase, these bands showed a slight red shift (267, 255, and 249 nm) and increased oscillator strengths, indicating enhanced charge-transfer character and solvent stabilization of the excited states (table 4). These results confirm strong intramolecular charge transfer (ICT) and improved optical responsiveness of NMBA in polar environments, supporting its potential for NLO and bioactive applications.
 |
Figure 6: Theoretical UV spectra of title compound in gas and water phase. Click here to View Figure |
Non-linear optical (NLO) analysis
Nonlinear optical (NLO) studies are fundamental in assessing molecular systems for their potential in photonic and optoelectronic technologies, with dipole moment, polarizability, and first-order hyperpolarizability serving as key descriptors.
The following expression is employed in NLO analysis:

Other equations used in this study are:

![]()
Where μ = the total static dipole moment, α = mean polarizability, and βtot = mean first-order hyperpolarizability.
Table 5: NLO properties for title compound in gas phase.
| NLO properties | Gas | Water | NLO properties | Gas | Water |
| Dipole moment (μ) Debye | Hyperpolarizability (β)x 10-33 e.s.u. | ||||
| μx | 1.1698 | 1.7394 | βxxx | -2328.3788 | -4833.1015 |
| μy | -0.6663 | -1.1840 | βxyy | 54.6339 | -1713.4862 |
| μz | -0.4250 | -0.7963 | βxzz | 136.0705 | -235.6226 |
| μ | 1.4117 | 2.2498 | βyyy | 4664.2689 | 16043.9889 |
| Polarizability (α)x10-24 e.s.u. | βxxy | 890.4217 | 2856.9939 | ||
| αxx | 45.7763 | 56.6409 | βyzz | -102.7649 | 381.0059 |
| αyy | 42.4766 | 60.4935 | βzzz | -174.8664 | -298.2945 |
| αzz | 24.8800 | 35.7608 | βxxz | 91.0015 | 302.4188 |
| < α > | 37.7110 | 50.9651 | βyyz | 67.4351 | 548.7870 |
| ∆α | 27.5170 | 32.5965 | βtot | 5856.0579 | 20447.4735 |
The nonlinear optical (NLO) properties of NMBA, calculated at the B3LYP/6-311++G(d,p) level (Table 5), were compared with those of urea, a standard NLO reference material [36]. The dipole moment (μ) of NMBA increased from 1.4117 D (gas) to 2.2498 D (water), whereas urea exhibits only 1.373 D, indicating that NMBA possesses greater molecular polarity and charge separation, particularly in polar media. The average polarizability (⟨α⟩) of NMBA was found to be 37.7110×10-24 e.s.u. (gas) and 50.9651×10-24 e.s.u. (water), approximately 8.3 and 11.3 times higher than that of urea (4.52×10-24 e.s.u.), signifying enhanced electronic cloud deformation under an applied electric field. Furthermore, the total first-order hyperpolarizability (βtot) of NMBA exhibited a remarkable increase from 5856.0579×10-33 e.s.u. (gas) to 20447.4735×10-33 e.s.u. (water), which is about 17.6 and 61.4 times greater than that of urea (333×10-33 e.s.u.), respectively. This substantial enhancement demonstrates the strong intramolecular charge transfer (ICT) between the aniline donor and benzoxazole acceptor units. Collectively, the higher μ, ⟨α⟩, and βtot values confirm that NMBA possesses far superior NLO characteristics than urea, underscoring its promise for advanced optoelectronic and photonic applications.
 |
Figure 7: Ramachandran plot of 1DQ9. Click here to View Figure |
Table 6: Molecular docking of the title compounds with anti-cholesterol target proteins.
| PDB ID | Binding energy Kcal/mol | Amino Acids | Distance (Å) | Type |
| 1DQ9 | -7.50 | C:ASN658 | 2.02428 | Conventional H Bond |
| C:PHE628 | 4.26212 | Pi-Pi Stacked | ||
| C:MET655 | 5.19079 | Alkyl | ||
| C:ALA654 | 4.28616 | Pi-Alkyl | ||
| C:MET655 | 5.11206 | Pi-Alkyl | ||
| C:MET655 | 4.26164 | Pi-Alkyl | ||
| C:ALA654 | 4.22481 | Pi-Alkyl | ||
| C:VAL805 | 4.63466 | Pi-Alkyl | ||
| C:ALA826 | 5.17370 | Pi-Alkyl |
Ramachandran plot
The Ramachandran plot of the 1DQ9 protein confirms excellent stereochemical quality, with most residues located within the energetically favoured regions (figure 7). The φ–ψ torsion angles are densely clustered in the α-helical (φ ≈ -60°, ψ ≈ –45°) and β-sheet (φ ≈ -135°, ψ ≈ 135°) regions, indicating well-defined secondary structures. Nearly all residues fall within the allowed and generously allowed regions, signifying minimal steric hindrance and optimal backbone geometry. Only a few residues appear in disallowed regions, likely representing flexible loops or functionally relevant conformations. Overall, the plot validates the structural reliability and stability of the 1DQ9 protein model for subsequent computational and docking analyses.
 |
Figure 8: (A) 3D visualization of ligand binding within the 1DQ9 active site; (B) 3D interaction; (C) 2D interaction; (D) aromatic surface; Click here to View Figure |
Molecular docking
Molecular docking was employed to explore the binding interactions of NMBA with the anti-cholesterol target protein ACAT (PDB ID: 1DQ9). The compound exhibited a strong binding affinity with an energy of -7.50 kcal/mol, indicating a stable and favourable interaction within the active site (Table 6). A key hydrogen bond with ASN658 (2.02428 Å) anchored the ligand, while π-π stacking with PHE628 and multiple hydrophobic contacts with MET655, ALA654, VAL805, and ALA826 (4.22481-5.19079 Å) further stabilized the complex. These interactions reflect effective accommodation of NMBA within the receptor pocket through electrostatic and van der Waals forces. Overall, the docking results highlight NMBA’s promising inhibitory potential against ACAT, supporting its candidature as a potential antihyperlipidemic agent and emphasizing the utility of molecular docking in rational drug design.
Figure 8 presents the molecular docking visualization of the NMBA-1DQ9 complex. The overall 3D structure and binding pocket are shown in Figure 8A, with an enlarged view in figure 8B highlighting key ligand-residue interactions. The 2D interaction diagram (Figure 8C) confirms a conventional hydrogen bond with ASN658 and multiple π-π and π-alkyl interactions involving PHE628, MET655, ALA654, VAL805, and ALA826, which collectively stabilize the complex. Surface analyses in Figures 8D-I depict aromatic, hydrogen-bonding, electrostatic, ionizability, solvent-accessible, and hydrophobic features of the binding site. These visualizations demonstrate that NMBA fits snugly within the active site, forming a stable and energetically favourable complex through strong hydrogen bonding and hydrophobic interactions, underscoring its potential as a promising anti-cholesterol agent.
Conclusion
In summary, the combined DFT and molecular docking analysis of NMBA reveals its promising structural, electronic, and biological features. Solvent polarity enhances its stability and charge delocalization, while FMO and DOS analyses indicate a moderate energy gap favourable for charge transfer. TD-DFT results show solvent-induced red shifts, confirming excited-state stabilization. Notably, NMBA exhibits a hyperpolarizability over sixty times that of urea, highlighting its strong NLO potential. Docking studies with 1DQ9 further demonstrate a stable binding energy (-7.50 kcal/mol) through hydrogen bonding, π-π stacking, and hydrophobic interactions, supporting its role as an anti-cholesterol agent. Overall, NMBA emerges as a multifunctional molecule with both optoelectronic and pharmacological potential.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Marati, K., Palatheeya, S., Chettupalli, A.K. et al.Characterization and interactions between piperine and ezetimibe in their Anti-hyperlipidemic efficacy using Biopharmaceutics and Pharmacokinetics. BMC Pharmacol Toxicol 26, 7 (2025). https://doi.org/10.1186/s40360-025-00836-z
CrossRef - Alloubani, A.; Nimer, R.; Samara, R. Relationship between Hyperlipidemia, Cardiovascular Disease andStroke: A Systematic Review. Current Cardiology Reviews 2020, 17 (6), 52 – 66. https://doi.org/10.2174/1573403×16999201210200342.
CrossRef - Hai, Q.; Smith, J. D. Acyl-Coenzyme A: Cholesterol acyltransferase (ACAT) in cholesterol metabolism: From its discovery to clinical trials and the Genomics era. Metabolites 2021, 11 (8), 543. https://doi.org/10.3390/metabo11080543.
CrossRef - Laeeq S, Dubey V. Insilico Screening for Identification of Novel Acyl-CoA: Cholesterol Acyltransferase Inhibitors. NeuroQuantology, 20, 2557-2567 (2022) doi: 10.14704/nq.2022.20.8.NQ22276.
- Laeeq S, Dubey V. Computational prediction of ADMET properties of ACAT inhibitors for synthesis and pharmacological screening. International Journal of Health Sciences. 6, 2377–2388 (2022) https://org/10.53730/ijhs.v6nS7.11866
CrossRef - Laeeq S, Dubey V. Study of ACAT receptor with heterocyclic compounds for drug designing. Int J Pharm Sci Res, 13, 2638-47 (2022) doi: 10.13040/IJPSR.0975-8232.13(7).2638-47.
CrossRef - Sattar, R.; Mukhtar, R.; Atif, M.; Hasnain, M.; Irfan, A. Synthetic transformations and biological screening of benzoxazole derivatives: A review. Journal of Heterocyclic Chemistry 2020, 57 (5), 2079–2107. https://doi.org/10.1002/jhet.3944.
CrossRef - Wong, X. K.; Yeong, K. Y. A patent review on the current developments of benzoxazoles in drug discovery. ChemMedChem 2021, 16 (21), 3237–3262. https://doi.org/10.1002/cmdc.202100370.
CrossRef - Arulmurugan, S.; Kavitha, H. P.; Vennila, J. P. Review on the synthetic methods of biologically potent benzoxazole derivatives. Mini-Reviews in Organic Chemistry 2020, 18 (6), 769–785. https://doi.org/10.2174/1570193×17999201020231359.
CrossRef - Di Martino, S., De Rosa, M. The Benzoxazole Heterocycle: A Comprehensive Review of the Most Recent Medicinal Chemistry Developments of Antiproliferative, Brain-Penetrant, and Anti-inflammatory Agents. Top Curr Chem (Z)382, 33 (2024). https://doi.org/10.1007/s41061-024-00477-6
CrossRef - Abou-Zied, O. K. Revealing the ionization ability of binding site I of human serum albumin using 2-(2′-hydroxyphenyl)benzoxazole as a pH sensitive probe. Physical Chemistry Chemical Physics 2012, 14 (8), 2832. https://doi.org/10.1039/c2cp23337a.
CrossRef - Abdullahi, A., Yeong, K.Y. Targeting disease with benzoxazoles: a comprehensive review of recent developments. Med Chem Res33, 406–438 (2024). https://doi.org/10.1007/s00044-024-03190-7
CrossRef - Algul, O.; Ersan, R. H.; Alagoz, M. A.; Duran, N.; Burmaoglu, S. An efficient synthesis of novel di-heterocyclic benzazole derivatives and evaluation of their antiproliferative activities. Journal of Biomolecular Structure and Dynamics 2020, 39 (18), 6926–6938. https://doi.org/10.1080/07391102.2020.1803966.
CrossRef - Mishra, V. R.; Ghanavatkar, C. W.; Mali, S. N.; Qureshi, S. I.; Chaudhari, H. K.; Sekar, N. Design, synthesis, antimicrobial activity and computational studies of novel azo linked substituted benzimidazole, benzoxazole and benzothiazole derivatives. Computational Biology and Chemistry 2019, 78, 330–337. https://doi.org/10.1016/j.compbiolchem.2019.01.003.
CrossRef - Rida, S.; Ashour, F.; Elhawash, S.; Elsemary, M.; Badr, M.; Shalaby, M. Synthesis of some novel benzoxazole derivatives as anticancer, anti-HIV-1 and antimicrobial agents. European Journal of Medicinal Chemistry 2005, 40 (9), 949–959. https://doi.org/10.1016/j.ejmech.2005.03.023.
CrossRef - Parlapalli, A.; Kumar, R. V.; Cidda, M.; Manda, S.; Synthesis, anti-inflammatory and antioxidant activity of n-(benzoxazol-2-yl)-2-(2-oxoindolin-3-ylidine)hydrazine carbothioamides. J Chem Pharm Res 9, 57 (2017).
- Reddy, N. B.; Burra, V. R.; Ravindranath, L. K.; Sreenivasulu, R.; Kumar, V. N. Synthesis and biological evaluation of benzoxazole fused combretastatin derivatives as anticancer agents. Monatshefte Für Chemie – Chemical Monthly 2016, 147 (3), 593–598. https://doi.org/10.1007/s00706-016-1685-y.
CrossRef - Kaur, A.; Pathak, D. P.; Sharma, V.; Narasimhan, B.; Sharma, P.; Mathur, R.; Wakode, S. Synthesis, biological evaluation and docking study of N-(2-(3,4,5-trimethoxybenzyl)benzoxazole-5-yl) benzamide derivatives as selective COX-2 inhibitor and anti-inflammatory agents. Bioorganic Chemistry 2018, 81, 191–202. https://doi.org/10.1016/j.bioorg.2018.07.007.
CrossRef - Arulmurugan, S.; Kavitha, H. P. Synthesis and potential cytotoxic activity of some new benzoxazoles, imidazoles, benzimidazoles and tetrazoles. Acta Pharmaceutica 2013, 63 (2), 253–264. https://doi.org/10.2478/acph-2013-0018.
CrossRef - Peng, J.; Liu, Y.; Yang, J.; Chen, Z.; Wang, K.; Li, A. Multifunctional elastic benzoxazole derivative crystals for advanced optoelectronic applications. Science China Materials 2024, 68 (1), 141–148. https://doi.org/10.1007/s40843-024-3140-4.
CrossRef - Barłóg, M.; Podiyanachari, S. K.; Bazzi, H. S.; Al‐Hashimi, M. Advances in Π‐Conjugated benzothiazole and Benzoxazole‐Boron complexes: Exploring optical and biomaterial applications. Macromolecular Rapid Communications 2025, 46 (8), e2400914. https://doi.org/10.1002/marc.202400914.
CrossRef - Rivera, E.; Ceballo, R.; Neira, O.; Avila, O.; Fonseca, R. DFT study of functionalized Benzoxazole-Based D–Π–A architectures: Influence of ionic fragments on optical properties and their potential in OLED and solar cell devices. Molecules 2025, 30 (18), 3737. https://doi.org/10.3390/molecules30183737.
CrossRef - Raftani, M.; Abram, T.; Azaid, A.; Kacimi, R.; Bennani, M. N.; Bouachrine, M. Theoretical design of new organic compounds based on diketopyrrolopyrrole and phenyl for organic bulk heterojunction solar cell applications: DFT and TD-DFT study. Materials Today Proceedings 2021, 45, 7334–7343. https://doi.org/10.1016/j.matpr.2020.12.1228.
CrossRef - Menon, V. V.; Mary, Y. S.; Mary, Y. S.; Panicker, C. Y.; Bielenica, A.; Armaković, S.; Armaković, S. J.; Van Alsenoy, C. Combined spectroscopic, DFT, TD-DFT and MD study of newly synthesized thiourea derivative. Journal of Molecular Structure 2017, 1155, 184–195. https://doi.org/10.1016/j.molstruc.2017.10.093.
CrossRef - Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JAJr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT.
- Barbieri PL, Fantin PA, Jorge FE (2006) Gaussian basis sets of triple and quadruple zeta valence quality for correlated wave functions. Mol Phys 104:2945-2954. doi: 10.1080/00268970600899018.
CrossRef - Jacquemin D, Perpète EA, Ciofini I, Adamo C, Valero R, Zhao Y, Truhlar DG (2010) On the performances of the M06 family of density functionals for electronic excitation energies. J Chem Theory Comput 6:2071-2085. doi: 10.1021/ct100119e.
CrossRef - Dennington, Roy, Todd A. Keith, John M. Millam (2016) “GaussView 6.0. 16.” Semichem Inc.: Shawnee Mission, KS, USA, 143-150.
- Zhurko, G. A., & Zhurko, D. A. Chemcraft – Graphical program for visualization of quantum chemistry computations.http://www.chemcraftprog.com/download.html
- Ram, A.; Chaturvedi, A. K.; Kishore, R.; Singh, R. N.; Srivastava, N.; Rawat, P.; Chaturvedi, D. Synthesis, computational studies, anticancer evaluation, and ADMET profiling of 2-thioxoimidazolidine-4,5-diones catalyzed by tetra-butyl ammonium iodide. Journal of Molecular Structure 2025, 1340, 142514. https://doi.org/10.1016/j.molstruc.2025.142514.
CrossRef - https://www.rcsb.org/structure/1DQ9
- BIOVIA, Dassault Systèmes, Discovery Studio, 2025, San Diego: Dassault Systèmes.
- Shashi R, Begum NS, Panday AK (2021) A rapid ultrasound synthesis of xanthenediones catalyzed by boric acid in ethanol-water medium: single crystal, DFT and Hirshfeld surface analysis of two representative compounds. J Mol Struct 1228:129794. doi: 10.1016/j.molstruc.2020.129794
CrossRef - Shashi R, Begum NS, Panday AK (2020) Green synthesis of tetraketones: crystal structure, DFT studies and Hirshfeld surface analysis of 2,2′-((3,4-dimethoxyphenyl) methylene) bis(3-hydroxy-5,5-dimethylcyclohex2-enone). Mol Cryst Liq Cryst 709:81-97. doi: 10.1080/15421406.2020.1829308
CrossRef - Shashi R, Begum NS, Panday AK, Foro S (2021) Green approach for the synthesis of xanthenedione derivatives: crystal structure, DFT, and Hirshfeld surface analysis. Mol Cryst Liq Cryst 730:35-51. doi: 10.1080/15421406.2021.1946350.
CrossRef - Parakkal, S. C.; Datta, R.; Saral, A.; Muthu, S.; Irfan, A.; Jeelani, A. Solvent polarity, structural and electronic properties with different solvents and biological studies of 3,3,5-triphenylfuran-2(3H)-one- cancers of the blood cells. Journal of Molecular Liquids 2022, 368, 120674. https://doi.org/10.1016/j.molliq.2022.120674.
CrossRef
Accepted on: 28 Nov 2025







ISSN Online: 2231-5039

![]()




{kind=link}