A Comprehensive Review on Chromene Derivatives: Potent Anti-Cancer Drug Development and Therapeutic Potential

Department of Physical Sciences, Chemistry Division, College of Science, Jazan University, Jazan, Kingdom of Saudi Arabia.
Corresponding Author E-mail: hmhamed@jazanu.edu.sa
DOI : http://dx.doi.org/10.13005/ojc/410229
Download this article as:
![]()
Chromene derivatives are a notable category of naturally occurring chemicals recognized for their varied pharmacological attributes. These heterocyclic structures demonstrate the capacity to engage with diverse cellular targets, enhancing their wide range of biological activities. Compounds derived from chromene exhibit antitumor, antioxidant, anti-inflammatory, anticoagulant, anticonvulsant, diuretic, hepatoprotective, antispasmodic, antifungal, antimicrobial, estrogenic, antiviral, anthelmintic, anti-HIV, antitubercular, hypothermic, herbicidal, vasodilatory, and analgesic properties. Their promise in cancer therapy has spurred the creation of new derivatives with improved therapeutic effectiveness. Structure-activity relationship (SAR) studies indicate that alterations to the chromene core, by targeted functionalization, can markedly improve their bioactivity. The review presents an overview of the anticancer effects of chromene derivatives, emphasizing recent developments in their structural alterations and modes of action.
KEYWORDS:Anti-cancer activity; 2H-chromene; 4H-chromene; One-pot three-component synthesis
Introduction
Worldwide, cancer is a significant public health issue. According to incidence and death statistics, it is rising in developing and developed nations. Considering advances in diagnosis and treatment, cancer remains to be the 2nd foremost reason of death internationally, after cardiovascular disease. Lung cancer exhibits the greatest fatality rates across all cancer forms in both genders. Conversely, breast cancer ranks as the second foremost reason for mortality associated with cancer in women. Despite numerous cancer treatment methods, research into new chemotherapeutic drugs continues. Because cancer is complicated and malignant cells are drug-resistant1, breast cancer kills many women worldwide. Elevated 17β-estradiol, a female sex hormone, causes breast cancer2. Multidrug resistance (MDR) is a key cause for therapeutic failure in the studies of cancer. Resistance to a medicine can be inherited or acquired; combination medications have been tried to address these hurdles. New medication design materials are still needed to overcome this issue3.
Chemotherapeutic drugs generally target rapidly reproducing cells; nevertheless, usually lack selectivity, impacting both malignant and healthy tissues. Although significant advances in cancer prevention and treatment, traditional chemotherapeutic agents including 5-fluorouracil, paclitaxel, cisplatin, and docetaxel are still extensively utilized, while cancer persists as a severe health concern4. The continuous pursuit of new anticancer medicines and innovative treatment methodologies constitutes a dynamic research domain, motivated by the necessity to discover novel biological targets and create therapies that exhibit superior efficacy, enhanced selectivity, diminished toxicity, and minimal adverse effects5.
Heterocyclic compounds are of significant importance in the field of drug development, as evidenced by the fact that over 70% of therapeutic drug compounds currently documented in the previous studies belong to this category. The oxygen-containing heterocycles stand out among these abundant heterocyclic compounds due to their significant prevalence in nature and their wide-ranging biological and pharmacological significance. The “chromene” heterocyclic scaffolds found in these O-heterocycles are notable for their biological activity6, 7.
The chromene unit is found in many different kinds of organic compounds. 4H-chromenes have a potent cytotoxic impact against a variety of human cancer cell lines through a variety of different mechanisms. These pathways contains depolarization of microtubules and disturbance of the tumor vasculature. CrolibulinTM (EPC2407), a chromene analog, is now participating in clinical trials to treat advanced solid malignancies. These trials are in Phase I/II8. Many scientists are looking for novel chromenes because of the promising anti-cancer effects of a number of chromenes found in medicinal plants. Various biological activity evaluations are attributed to chromenes, including anti-cancer, antibacterial, antiviral, anti-inflammatory, antipyretic, and ulcerogenic9-12.
The benzene ring of chromenes is fused to a pyran nucleus, making them a heterocyclic chemical. In 2H-chromenes I, C-2 forms a double bond with oxygen (2-oxo-2H-chromene, also known as coumarin), (2-imino-2H-chromene), inside the 2H-chromene-2-thione structure may establish a single bond with two different substituents (X, Y) or engage in alternative bonding arrangements. In 4H-chromene derivatives (III and IV), the carbon at the 4-position can either exhibit sp³ hybridization or form a double bond with a heteroatom, hence affecting the compound’s structural and electrical characteristics. The 4H-chromones are particularly important, especially if the 2-position contains an aromatic ring. These later chemicals, usually referred to as flavones, are frequently found in normal goods, and the existence of this structure in dietary products has been linked to a number of health benefits, thus elevating the significance of this nucleus13.
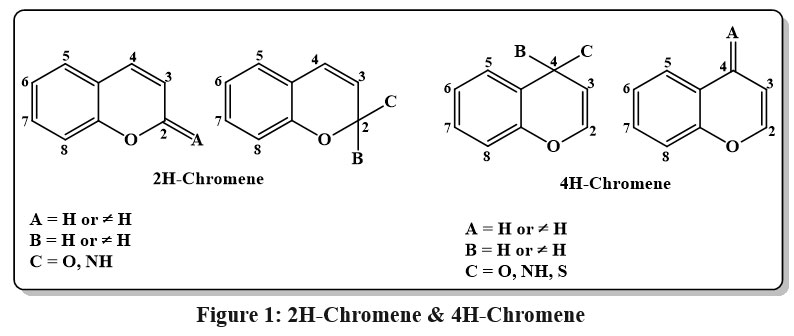 |
Figure 1: 2H-Chromene and 4H-Chromene |
This review offers a comprehensive and current overview of synthetic methodologies, reaction systems, biological activities, and structure-activity relationships (SAR) concerning the nucleus of 2H/4H-chromene and its implication in the progress of bioactive compounds, by addressing the absence of recent consolidated updates. This review seeks to aid medicinal chemists and researchers in the creation and optimization of innovative 2H/4H-chromene-based compounds for the treatment of serios medical condition and possible industrial applications.
Nature-based anti-cancerous chromene derivatives
Mallotus apelta (Lour.) Müll.Arg has been utilized for the handling of chronic hepatitis in traditional Chinese medicine for centuries. Seven compounds (including six novel chromene based derivatives called malloapeltas C-H and one previously recognized molecule called malloapelta B) have been isolated and described from the plant’s leaves. The TOV-21G ovarian cancerous lines cell line was utilized in a cell count kit-8 (CCK-8) experiment to assess the cytotoxicity of each medication. Six medicines exhibited notable their inhibitory impact on expansion and persistence on the TOV-21G ovarian cancer cell line, with GI50 rates extending between 0.06-10.39 μM and the value of IC50 between 1.62-10.42 μM. The mechanism of apoptosis was investigated using three of the most cytotoxic compounds. The stimulation of caspase 8, 9, and PARP, along with the overexpression of Bax and Bak and a reduction of Bcl-xL and enduring, served as indicators that compounds 1a & 1b, 2, and 3 (Fig. 2) triggered apoptosis. Additional research revealed that compounds 2, 3 effectively suppressed the NF-κB signaling pathway12.
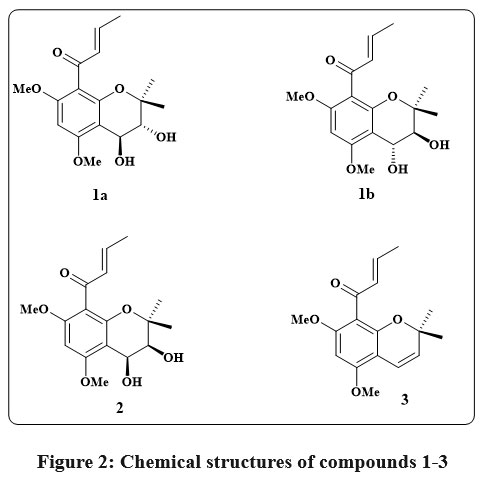 |
Figure 2: Chemical structures of compounds 1-3 |
Fig. 3 shows the chromene compound psoralidin 4, which occurs naturally in Psoralea corylifolia. This compound’s potent cytotoxic effects against numerous cancerous lines of cell, containing those of the gastric, colon, and breast, led to its identification as a promising anticancer agent14, 15. As a small component like of Neo-tanshinlactone 5 (Fig. 3) was separated from the ethanolic root extract of Salvia miltiorrhiza. The anticancer properties of this chromene were evaluated against several therapeutically relevant patient breast cancer cell types. The findings indicated low ED50 ranges 0.20 to 0.60 µg/mL for both estrogen receptor-positive (ER+) lines of cell, like MCF-7 receptor and ZR-75-1 receptor, as well as for one ER-/HER-2 carrying cell line, SK-BR-316, 17. Additionally, the marine organism Aplidium conicum was obtained off the coast of Sardinia, Italy, to yield the thiaplidiaquinones A 6 and B 7 Fig. 3. Using a human leukemia T line cell, researchers examined the impact of these complex compounds on the survival of cell. Additionally, the technique of flow cytometry and propidium iodide (PI) stains allowed the observation of a dramatic decrease in nuclear DNA in the treated cells. Reactive oxygen species (ROS) were also produced in high quantities by the cells in response to both compounds. Therefore, induction of cell death via the apoptotic pathway was inferred for Thiaplidiaquinones 6 and 718.
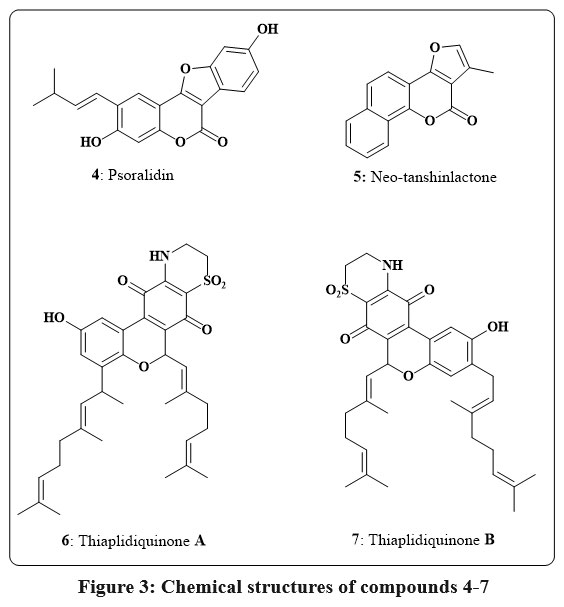 |
Figure 3: Chemical structures of compounds 4-7 |
The Coumarins 8 (Fig. 4), derived from the Mammea americana plant found in tropical and subtropical regions, was discovered to hinder the induction of hypoxia-inducible factor-1 (HIF-1) in the prostate and the breast cancerous lines of cell. These specific dihydroxy-coumarins, possessing isoprenyl groups, exhibited notable potency with EC50 values mostly below 10 µM for the breast cancer cells like T47D as well as for the prostate cancer cell line like PC-3. Coumarins featuring a 6-prenyl-8-(3-methyloxobutyl) components explained the heightened effectiveness in inhibiting HIF-119. Two different compounds, 9 and 10 (Fig. 4), were extracted from the fruit as well as from the bark of stem of the Calophyllum plant, found primarily in tropical rain forests. They were checked for their cytotoxicity employing the line cell KB, and the results showed that at concentrations ranging from 4-8 µg/mL, they inhibited the growth of the KB cells by up to 50%20.
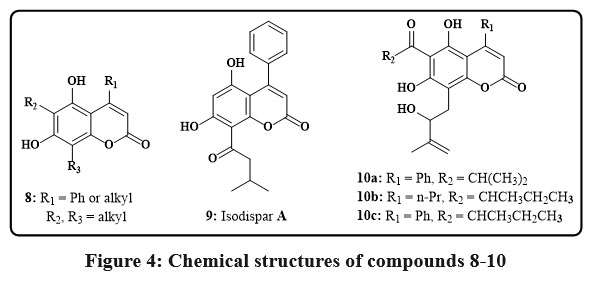 |
Figure 4: Chemical structures of compounds 8-10 |
In the Egyptian plant Polygonum senegalense, the fungus Alternaria species produced several natural compounds known as chromenes 11a-e, as seen in Fig. 5. The cytotoxic efficacy of these agents versus L5178Y murine lymphoma cells was evaluated. In particular, compounds 11a-c showed significant efficacy, with EC50 ranging from 1.7-7.8 µg/mL. Additionally, 24 distinct protein kinases were tested to determine these isolated chromenes’ in vitro inhibitory capability. These substances successfully inhibited several enzymes, according to the study’s findings, and they showed promising IC50 values of less than 10 µg/mL21, 22. The Kielmeyera albopunctata plant is native to the Brazilian plateau and can grow freely. Two compounds, 12a and 12b (Fig. 5), were separated from the dichloromethane stem bark that is the extract of the plant and tested for their cytotoxicity. These drugs shown anticancer efficacy against multiple human cancer cell lines, including KB is the verbal epidermoid carcinoma, prostate cancer from LNCaP, and lung cancer from Lu1 and colon cancer from Col2, with an ED50 ranging from 11.1 – 19.8 µg/mL23. Compounds based on chromene have been seperated from the fresh leaves of the Marila pluricostata tree, endemic to Panama, Colombia, and Costa Rica. Compounds 13a, 13b, and 16 were found to be novel compounds, and chromenes 14a-c and 15a-c were known derivatives. Anticancer activity was shown by this group of chemicals in the cancer lines cell GI50, MCF-7, SF-268, and with H-480 values are very low like of 0.1 µg/mL24. The effectiveness of the naturally occuring coumarins 17a–c (Fig. 5) was obtained regarding their capacity to cleave DNA. This judgment was made in light of the fact that substances that can damage DNA strands have shown potential as anti-tumor therapies. The roots of Mallotus resinosus were used to produce these particular compounds. Although they were not particularly successful in causing DNA cleavage, the researchers noted that these substances showed potential as building blocks for synthetic derivatives25.
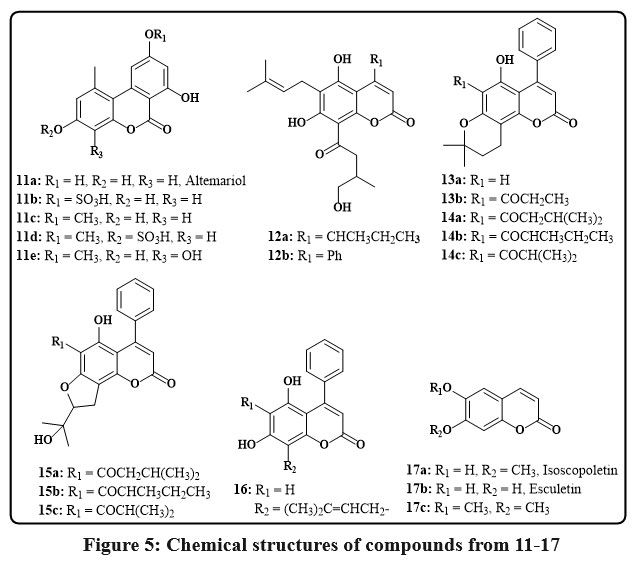 |
Figure 5: Chemical structures of compounds from 11-17. |
The leaves of the Euphorbiaceae plant Mallotus apelta Muell.-Arg. yielded two novel benzopyrans Fig. 6, designated 18 and 19. Detailed spectroscopic analyses were used to infer their molecular structures, emphasizing one and two dimentional techniques like mass spectrometry (MS) as well as nuclear magnetic resonance (NMR) data. In conjunction with an IC50 of 0.49 µg/mL versus human hepatocellular carcinoma represented by Hep-2 and 0.540 µg per mL against rhabdomyosarcoma (RD), Compound 18 displayed potent cytotoxic effects in the realm of biological activity. Additionally, compound 19 demonstrated an average activity level toward the Hep-2 cell line, having an alternative value of IC50 is of 4.22 µg/mL, as determined through in vitro assays26.
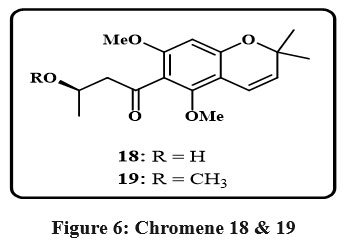 |
Figure 6: Chromene 18 and 19 |
Synthetic anti-cancerous chromene derivatives
Synthetic routes for anti-cancerous chromene derivatives
Numerous chromene derivatives with promising anticancer activity have been synthesized, with some exhibiting cytotoxic impacts on cancerous cell lines at different concentrations. The aminated nitrochromenes 23a-23b have been synthesized by using salicylic acid 20 and nitrostyrene 21 to get an intermediate nitrochromene 22 in the existance of DABCO which is 1,4-Diazabicyclo[2.2.2]octane at 40 ℃ temperature. The final product was obtained by oxidative Michael addition reaction of conjugate aniline with intermediate 22 in the existance of DMF (dimethyl formamide) and basic condition at room temperature Scheme 1. MTT assay assessed the cell growth inhibitory/cytotoxicity of 23a and 23b compounds on HepG2 (Human heptoma cells) and HEK293 (Human embryonic kidney cells). Compounds 23a and 23b were tested in HEK293 cells at 0-200 μM concentrations for 24 and 48 hours. In HepG2 cells, compounds D19 and D22 were tested at 0–100 μM concentrations during 48 hours. Cytotoxicity studies on HEK293 cells indicate that compounds 23a and 23b do not inhibit cell growth in the examined concentration range, instead for a small inhibition at 100 μM. Concentration-dependent cell growth inhibition by 23a and 23b compounds showed IC50 average values of 18.33 μM and 26.22 μM. These findings concluded that the compounds present 23a and 23b inhibit cancer cell multiplication without damaging normal cells27.
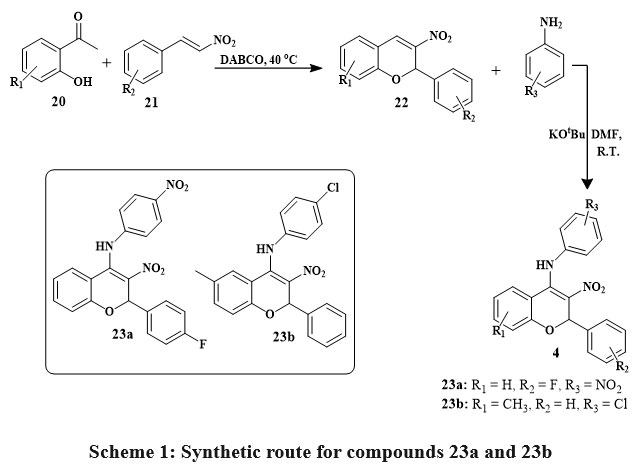 |
Scheme 1: Synthetic route for compounds 23a and 23b. |
Kabbe’s multi-component reaction was used to thermally condense cyclohexanone, pyrrolidine, and 1′-hydroxy-2′-acetonaphthone in methanol to yield compound 24. The methyl hydrazine-carbodithioate 26 joined with spirobenzo[h]chromenone compound 24 to yield intermediate 27. The S-methyl functional group was displaced during the condensation process of the middle products 26 with corresponding reflux of amines in isopropanol for 48 hours until methyl mercaptan changing stopped, yielding the final targeted thiosemicarbazide compound 28 Scheme 2. The precursors materials 29a and 29b were synthesized utilizing Kabbe’s multicomponent process. The compounds 29a, and b react with hydrated hydrazine to form hydrazone derivatives 30a,b. Hydrazone derivatives coupled directly with various phenyl isocyanates to provide the definitive tailored semi carbazide molecules 31a-e Scheme 3. Similar to the preceding reaction, compound 24 reacts with hydrated hydrazine to produce the intermediate hydrazone chromene 32. In the existance of triethylamine, the middle products 32 was refluxed directly with adamantyl carbonyl chloride to produce the intended outcome 33 in high yield Scheme 428.
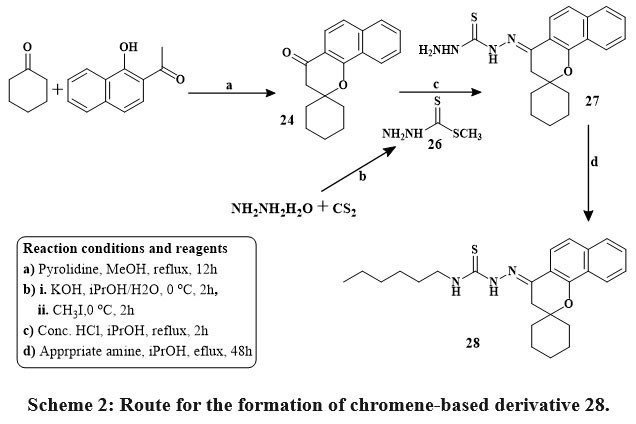 |
Scheme 2: Route for the formation of chromene-based derivative 28. |
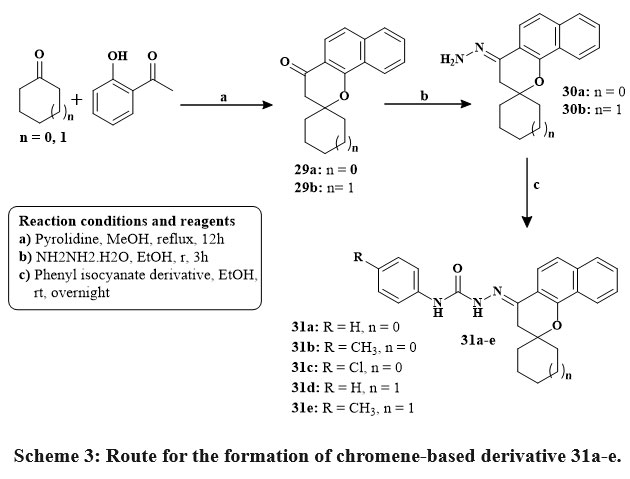 |
Scheme 3: Route for the formation of chromene-based derivative 31a-e. |
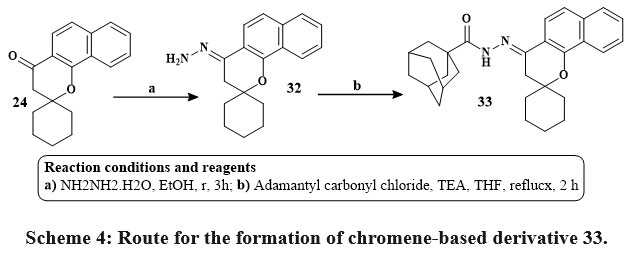 |
Scheme 4: Route for the formation of chromene-based derivative 33. |
Eight compounds (27, 28, 31a–e, and 33) exhibited significant anticancer action, with value of IC50 between 1.78 and 5.47 µM, surpassing those of sorafenib (between 3.6 and 6.2 µM) and erlotinib (IC50 more than 20 µM). A mechanistic investigation was conducted on selected compounds 28, 31c, and 33 against key anticancer targets, including B-RAF, EGFR, and tubulin. These compounds demonstrated stronger EGFR binding affinity than B-RAF kinase. Compounds 28, 31c, and 33 have comparable IC50 values to erlotinib for EGFR (1.4, 1.9, and 1.2 µM) and B-RAF kinase (2.7, 2.9, and 2.6 µM). Among them, compound 33, a hydrazine derivative of spiro-benzo-chromene, exhibited the highest potency against EGFR (IC50 = 1.2 µM), indicating an optimal steric and electronic configuration for kinase inhibition. Additionally, compounds 28, 31c, and 33 moderately inhibited tubulin polymerization. Docking simulations and in vitro assays support the potential of this spiro compound family as dual EGFR and B-RAF inhibitors, offering promising candidates for further anticancer drug development28. In the compound 6-, 7-, and 8-Carbon linear dicarboxylic acids was used to study the effects on the length of joiner on HDAC (histone deacetylases) inhibitory action. The amide (40) and ortho-aminoanilide (37–39) were employed as zinc-binding groups (ZBGs) in this series generated from dicarboxylic acids. The ortho-aminoanilide series’ interior cavity motifs, phenyl (38a-c) and 5-methylthiophen-2-yl (39a-c), are thought to enhanced the efficacy of inhibitory HDAC and selectivity of isoform. The 6-aminohexanoic acid as well as 5-amino pentanoic acid were utilized as linker motifs in addition to linear dicarboxylic acid, and the resulting ω-amides (42) and α-amino amides (43a, 43b) acted as ZBGs. Scheme 5 depicts the production of HDACi based on 8-ethoxy-3-nitro-2H-chromene29.
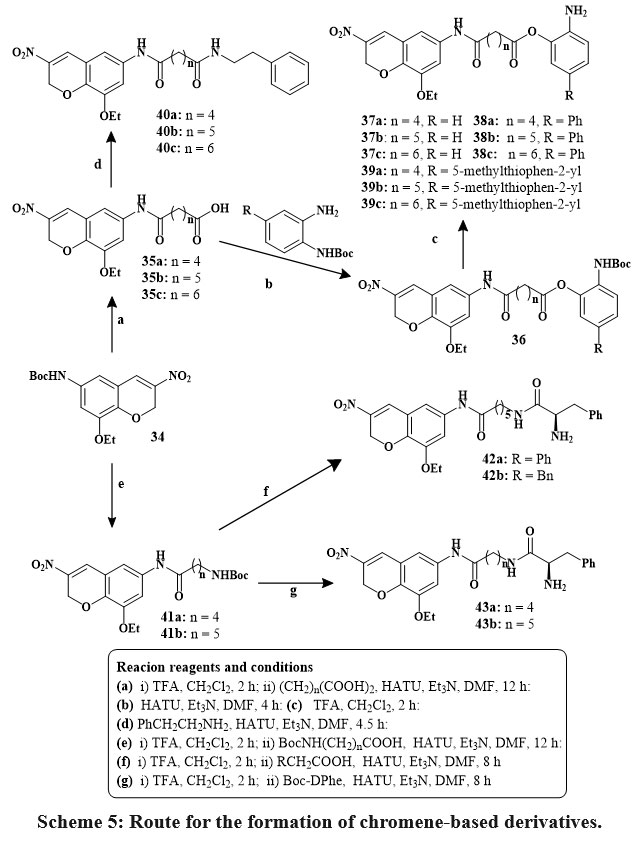 |
Scheme 5: Route for the formation of chromene-based derivatives. |
The inhibitory activity of a variety of HDACIs based on 8-ethoxy-3-nitro-2H-chromene against HDAC1, 2, and HDAC6 in MCF-7, A549, PC3, HeLa, and K562 cell lines was investigated. Compared to the basic drugs MS-275 and SAHA most coumarin derivatives exhibited stronger antiproliferative action teasted against the various five tumor lines cell. Strong HADC1 antagonists with IC50 amount in the nanomolar (nM) included compounds 37b, 37c, 38c, 39b, 43a and 43b. ZBG analogues (40a, 40b, 42) based on amides were high selective for HDAC2 and HDAC1. The phenyl internal cavity motif compounds 38b and 38c were more effective HADC1 inhibitors and selective over HADC2 than MS-275. D-Phe-derived compounds of ZBG were more effective HDAC inhibitors than their amide counterparts29.
Scheme 6 depicts the synthetic approaches taken to produce the desired molecules. Desired products 46a-j were obtained by reacting 4-chloro-1-naphthol 44 using α-cyano mono/di/tri substituted cinnamonitriles 45a-j in ethanolic piperidine, which during reflux for one hour. Among recently synthesized compounds, analogs 46b, e, c, i, h and 46d, j, f, a exhibited strong inhibition towards MCF-7. Notably, analogs 46b, 46a–j, especially 46e and 46h, showed high inhibition for HCT-116. In HepG-2, analogs 46e, h, b, 46c, d, j displayed significant inhibitory effects surpassing vinblastine and colchicine. Compound 46e outperformed doxorubicin against HepG-2, while others showed varying activity levels across cell lines. Structure-activity relationship suggests mono/dichloro-substituents at the position of para in the ring of phenyl in 4H-benzo[h]chromene enhances anticancer activity30, 31
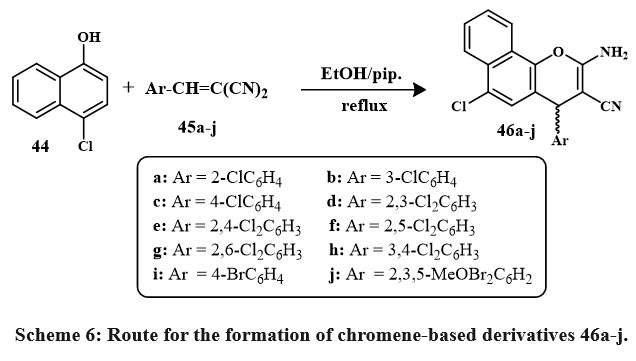 |
Scheme 6: Route for the formation of chromene-based derivatives 46a-j. |
Synthesized 4H-chromenes (51, 52, 55–57) and 5H-chromene 2,3-d-pyrimidines (53, 54, 58–60) Scheme 7 & 8 were examine versus HCT-116, A549, MCF-7, HepG-2 cancerous lines of cells. Some compounds 60a, 58a, 52b, 56a, 60b, 59b, 50b, and 58b inhibited MCF-7 cells strongly. 60b outperformed vinblastine and colchicine in HCT-116 anti-tumor effects. Vinblastine and colchicine were less effective than compounds 59b, 52b, 58a, 60a, b, 56a, 58b, 59a, 50b, 52a, 56b, 55a, and 57b against HepG-2. Compared to conventional medications, compounds 59b, 50a, 58a, 59a, 58b, 60b, a, 55a, 50b, 56a, and 57b showed promising anticancer activity against A549.
SAR studies represented that pyrimidine nuclei at the position of 2 and 3 with specific hydrophilic groups or hydrophobic groups and chromene nuclei at the 2-position or 7-position increased activity compared to other hydrophobic groups. Additionally, choosing 4-bromophenyl over 4-chlorophenyl at the position of 5- or 4- decreased the logarithm of the partitioning factor, Log P. In vitro, anticancer activity was higher in compounds with pyrimidine moieties and 4H-chromenes than in independent derivatives32.
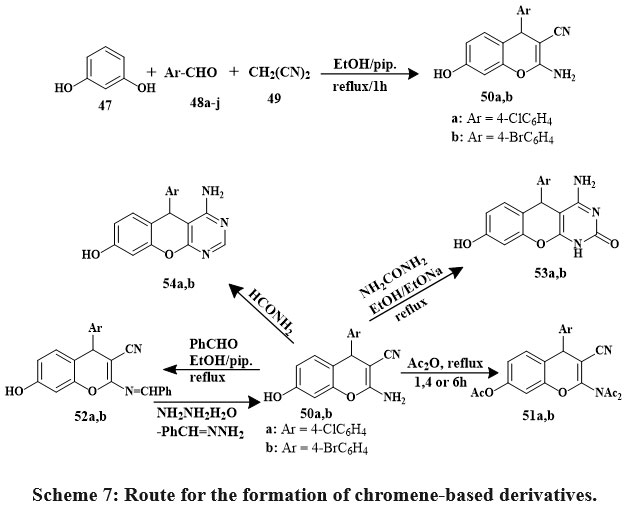 |
Scheme 7: Route for the formation of chromene-based derivatives. |
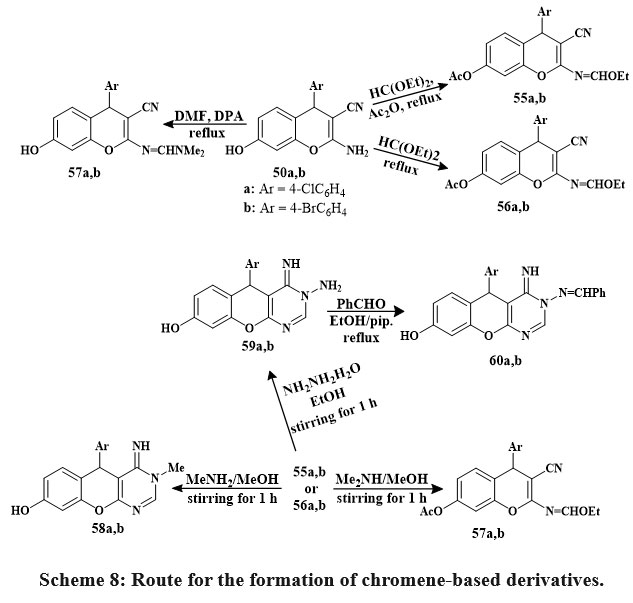 |
Scheme 8: Route for the formation of chromene-based derivatives. |
A new class of compounds was synthesized using Knoevenagel condensation and Michael addition adducts33. Two distinct chromene chemical families were generated using multi-component reactions (MCRs) to investigate their biological structure-activity relationship. The initial family resulted from the reaction between malononitrile 61, aromatic aldehyde 62a-i substituents, and 3-amino-2-naphthol 64 in the presence of piperidine. Refluxing the solutions for 2 hours led to compounds 65a-i with yields ranging from 60% to 77% (Scheme 9). The same synthetic approach was applied to produce the second set of chromene derivatives. By reacting malononitrile 1 with various aldehydes 63a-e and 5-amino-1-naphthol 66, compounds 67a-e were obtained. Three cell lines showed strong antiproliferative activity with IC50s of 0.3 to 2 µg/mL for compounds 65a, 65b, 65c, and 67c. For the most significant compounds, the enzyme activity assays were used for apoptotic study to determine their antiproliferative effects34.
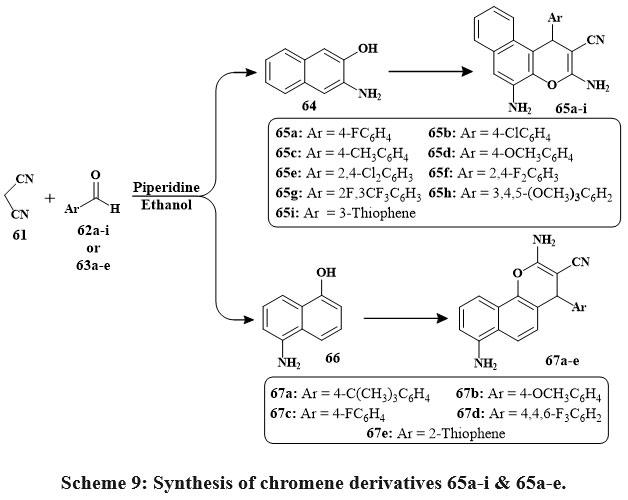 |
Scheme 9: Synthesis of chromene derivatives 65a-i & 65a-e. |
The preparation of conjugated imino chromenes 70a‑g and derivatives of phenyl-imino chromene 71a‑g is illustrated in Scheme 10. Various conjugated 2-imino chromenes 70a‑g were produced using the Knoevenagel synthesis of 2-cyano acetamide 68a‑g and 2-hydroxy benzaldehyde. Subsequently, subjecting these 2-imino chromenes to a elementary reaction involving aniline in reacted acetic acid led to the conversion into their corresponding phenyl-iminochromenes with good yields ranging from 70% to 85%. Phenyl-iminochromene derivatives 71a‑g with percentage I of 31 to 50.7 exhibited greater cytotoxic efficacy compared to their imino chromene equivalents at a concentration of 10 µM that is quivalent to %I of range of 25.1 to 36.3 versus the MOLT-4 lines of cells. Notably, compound 8g displayed remarkable inhibitory capacity of 51.7% against the MOLT-4 cell line at a dose of 10 µM35.
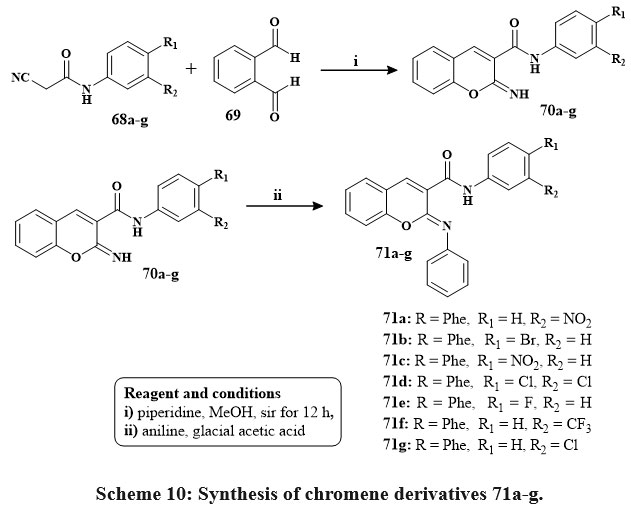 |
Scheme 10: Synthesis of chromene derivatives 71a-g. |
Compound 74 was prepared by reacting compound 72 with 1-phenylpiperidine 73 in the existance of solvent ethanol, and the mixture of reaction was refluxed for 1-15 hours Scheme 11. Their effectiveness toward cancerous lines of cells HeLa, – A549, – Hep2 and was assessed. Compound 74, identified as 6-methyl-3-nitro-4H-chromene-2-amine derivative substituted with a 4-(piperidin-1-yl)phenyl group at position 4, demonstrated the significant pronounced cytotoxic potential across each of the three cancerous lines cell (Hep2, A549, and HeLa), replacing IC50 amount from 0.75 ± 0.06, 4 ± 0.35, and 9 ± 0.73, respectively. Toxicity studies indicated the selective targeting of cancer cell lines by the 4-aryl-4H-chromenes36.
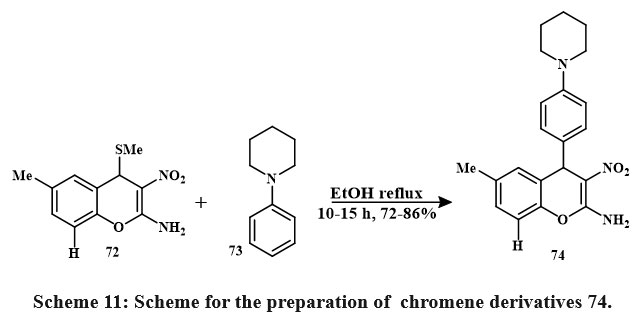 |
Scheme 11: Scheme for the preparation of chromene derivatives 74. |
Vilsmeier-Haack formylation of coumarin 75 yielded compound 76 Scheme 12. Compound 78a’s production is an example of the general process for synthesizing compounds 78a–c. The MTT tetrazolgtium reduction assay shown influenced by concentration cytotoxic impacts on tumor-derived human lines of cells within the micromolar level. The series’ strongest antiproliferative was Hydrazone 78a. In the leukemia HL-60 cell line, compound 78a has the most antiproliferative efficiency (IC50 2.9 μM), Subsequently, compounds 78c and 78b were examined. Antiproliferative efficacy in leukemia HL-60 cell lineage (IC50 2.9 ± 0.4 μM), followed by compounds 78c and 78b37.
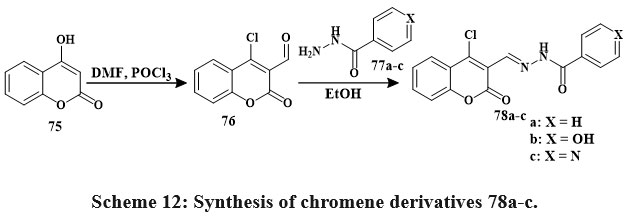 |
Scheme 12: Synthesis of chromene derivatives 78a-c. |
Treatment of a solution of 1-naphthol naphthol 81 in EtOH and piperidine with compounds 79a–79h or 84a–84g obtained in the formation of a new compound series Scheme 13. The mixture resulting from the reaction was heated while refluxing. After filtration, the solid products that were produced in this way were collected, washed with MeOH, and re-crystallized from ethyle alcohol or with thw benzene, respectively38.
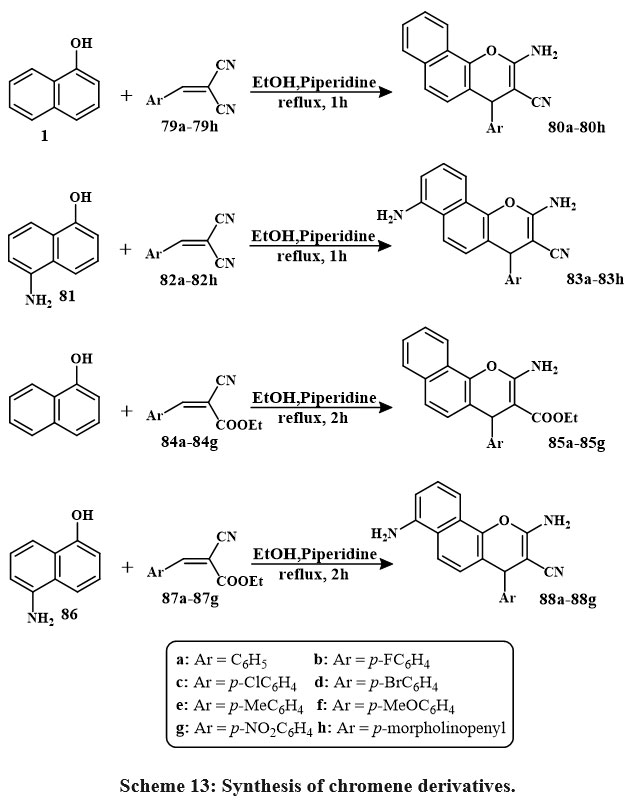 |
Scheme 13: Synthesis of chromene derivatives. |
The MTT colorimetric assay compared the synthesized compounds’ in-vitro cytotoxic efficacy toward HepG-2, MCF-7, and HCT-116, lines of cells to vin-blastine and colchicine. Anti-tumor assessment showed chemicals 88c, 85d, 85b, 85e, 88g (IC50 = 3.1 to 5.3 μg/mL) and 88c, 85d, 85b, 85e, 88g, 85c, 88e, 83f, 80a (IC50 = 3.1-17.5 μg/mL) reduced MCF-7 growth contras to vinblastine and colchicine. Compared to colchicine, 85b, 85d, 85e, 80a, 88b, 88a, 88c, 80d, 80g, 80f, 80b, 80h, 88b, 88a, 88e, 80c, 83g, 80e, 88g, 83f, 80f (IC50 = 2.9-32.2 μg/mL) decreased HCT-116 growth. Additionally, chemicals 85e, 80g, 80a, 85c (IC50 = 2.5-4.1 μg/mL) and 85e, 80g, 80a, 85c, 85b, 85a, 88f, 88e (IC50 = 2.5-10.4 μg/mL) decreased HepG-2 growth contras to vin-blastine and colchicine38. Compound 89 is the precursor for the production of novel heterocyclic substances. Consequently, the procedure of 1,4-Phenylenebis(2-cyanoacetamide), a dual-functional derivative of cyano-acetamide featuring a para-phenylene structure 89 with dimethylformamide-dimethylacetal (DMF and DMA) in mixing di-methylformamide, synthesizes enaminonitrile 90 Scheme 14.
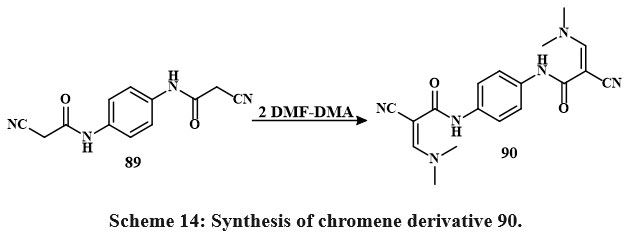 |
Scheme 14: Synthesis of chromene derivative 90. |
The chemical reactivity of enaminonitrile 90 has been examined with other phenolic and analogous chemicals, as depicted in Scheme 15. Upon refluxing chemical 90 with resorcinol in glacial acetic acid, a singular product, 91, was obtained. Similarly, its interaction with 2-nitroso-1-naphthol in the presence of zinc metal and glacial CH3COOH obtained in the synthesis of compound 92. Furthermore, the reaction of 90 with acid of thiobarbituric under reflux in the acid of glacial acetic yielded the pyrano-[2,3-d]-pyrimidine compound 93. Additionally, the reaction of 90 with thio-cyano-acetamide in N,N-dimethylformamide, catalyzed by triethylamine, yielded compound 9439.
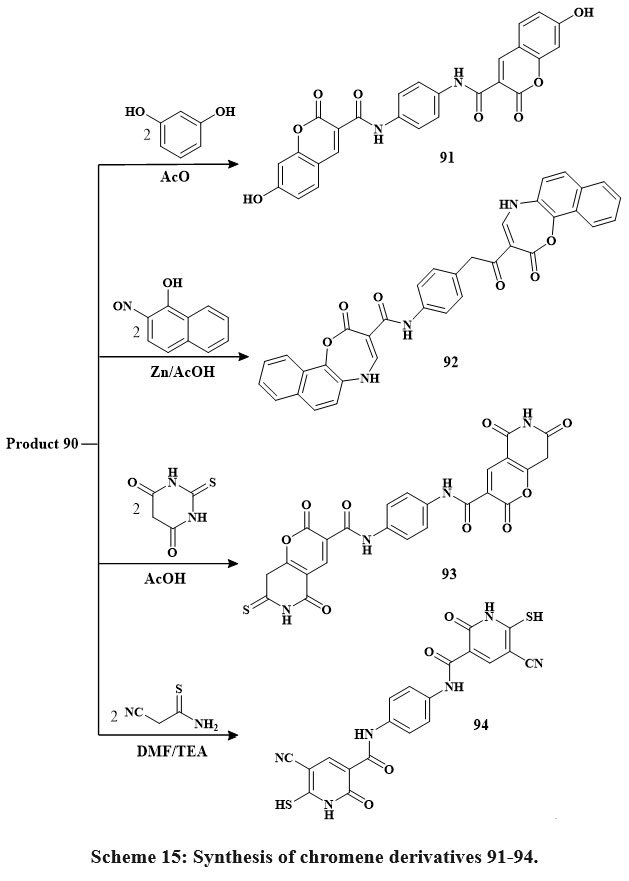 |
Scheme 15: Synthesis of chromene derivatives 91-94. |
The newly substances produced were assessed for their anticancer efficiency in vitro utilizing the standard MTT assay. The evaluation was performed using a set of four human cancerous lines cell: HepG-2 (hepatocellular carcinoma in the liver), HCT-116 (colorectal carcinoma in the colon), MCF-7 (mammary gland carcinoma in the breast), and Hep-2 (epidermoid carcinoma in the larynx). The anticancer screening showed that compounds had the highest in vitro cytotoxic activity relative to 5-fluorouracil. additionally, the IC50 amount range of the substances are as follows: HepG-2 (91, 92, and 93, IC50 = 2.41, 2.59 and 2.53 µg/mL), HCT-116 (91, 92, 93, and 94: IC50 = 4.98, 5.44, 5.32, and 5.20 µg/mL), MCF-7 (91, 92, 93, and 94: IC50 = 6.72, 6.99, 6.84, and 6.52 µg/mL). Further, the percentage of inhibition ranged from 3.47 to 4.06 g/mL for compounds 91, 92, 93, and 94 when tested versus the lines of cells in breast cancer MCF-7. Some substance like 91 was the prominent active material in this study, especially versus human Hep-2., HepG-2, and HCT-116 Compound 94 also was a prominent active material in this study, especially against MCF-739.
The active methine substituted 4H-chromene 97a-m was prepared by reacting active methylene 96a-m with C4-methanesulfanyl 4H-chromenes 95 (Schemes 16). Compared to the other solvents and bases that are currently using, it has been noted that in EtOH, the secondary amine L-proline refluxed is proved to be the most effective method for producing 97a-m as a pale yellow solid with quantitative yield. In the same manner, compound 99 was prepared using compound 98 and active methylene40.
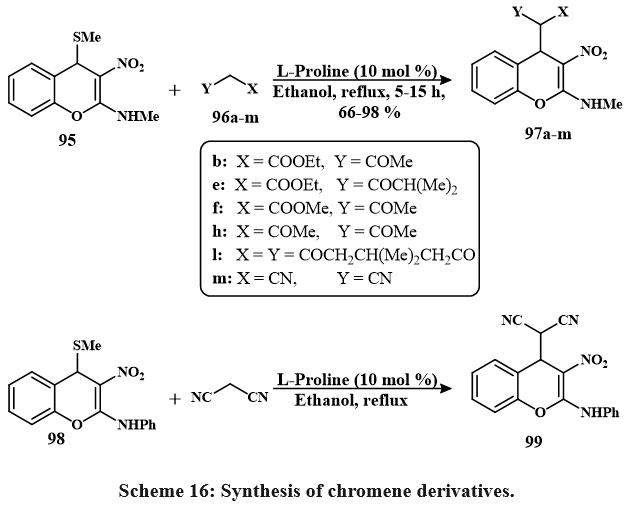 |
Scheme 16. Synthesis of chromene derivatives. |
 
The 4H-chromenes is a newly produced that were tested for their anti-cancerous characteristics by employing four different lines of cells. The results depicted that the 4H-chromenes with the C4-malononitrile conjugated were the highest effective at inducing programmed cell division. The results of the in silico investigations evaluated the combined relationship with the protein Bcl-2 matched the results of the in vitro studies. When taken together, the in vitro as well as in silico data proposed that the 4H-chromene 99 displayed IC50 values of HepG-2 (0.72 to 0.06 µM), A549 (1 to 0.08 µM), HeLa (0.7 to 0.05 µM), and HT-29 (0.85 to 0.07 µM), that this value is lead for the subsequent progress of more effective anti-cancerous compounds40.
In the presence of reflux with acetone and dehydrated K2CO3, benzils, and phenyl or 4-methoxyphenyl acetyl chlorides produced coumarins 100 in its unadulterated state through straightforward crystallization in high yields (90–95%). The target compounds were produced in high yields via base-catalyzed O-alkylation of the hydroxy group in compounds 100 using piperidine/pyrrolidine 1-(2-chloroethyl) monohydrochlorides 101 & 102 Scheme 17. Pure 6-aroylated chromenes 105 and 106 were crystallized in good yield. The chemical reaction of component 106 with piperidine or with pyrrolidine in dry DMF at 80 ℃ with TBAI produced alkylaminoalkoxy derivatives 107 and 108 Scheme 1841.
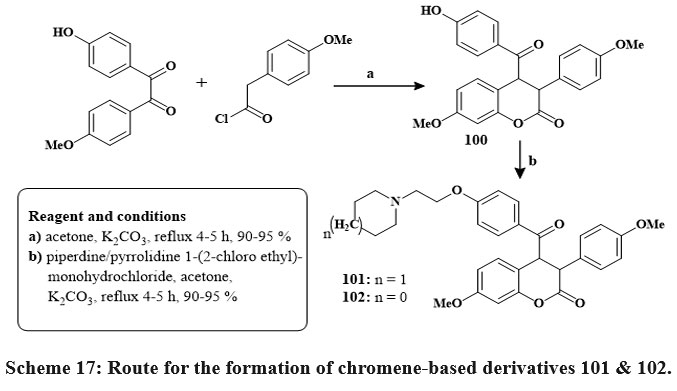 |
Scheme 17: Route for the formation of chromene-based derivatives 101 & 102. |
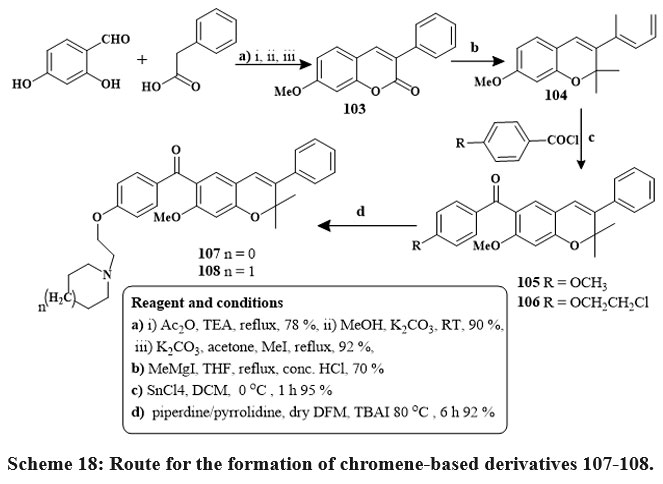 |
Scheme 18: Route for the formation of chromene-based derivatives 107-108. |
In the breast cancer cells, the compounds 101 and 102 had IC50 amount of 5.7 and 12.8 µM in MCF-7 and 18.4 and 12.7 µM in MDA-MB 231, comparable to raloxifene as well as tamoxifen standards. Compounds 107 and 108 inhibited MCF-7 cells at 6.8 µM to 8.1 µM efficacy41.
Scheme 19 depicts the synthetic route to produce the chromene derivatives 112a, b. By Henry-Knoevenagel condensation of various benzaldehydes 109 h, l, β-nitrostyrene’s 110a, b were created. High yields of the required 110a & b were produced with little to no by-product. In the presence of a base called (DABCO), 110a, b reacts with the suitable salicylaldehyde 111 to produce 112a, b, the corresponding final products. Compounds 112a and b were evaluated for cytotoxicity versus MDA-MB-231, T-47D, and MCF-7 lines cell of breast cancer. The compounds were more powerful than etoposide at cytotoxic activities versus chosen cell lines. The compound 112b illustrated considerable activity against MCF-7 cells, with an IC₅₀ of 0.2 µM, rendering it 36 times more potent than etoposide. The capacity of compounds 112a and 112b to trigger apoptosis was definitively established via acridine orange/ethidium bromide double labeling, TUNEL evaluation, and caspase-3 activation assays42.
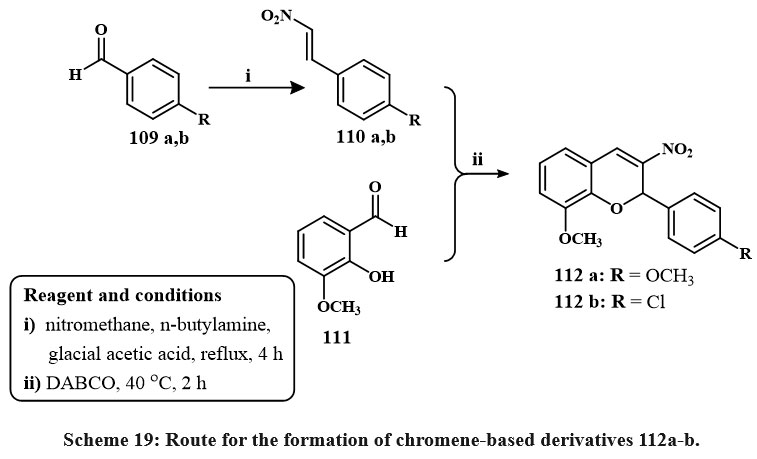 |
Scheme 19: Route for the formation of chromene-based derivatives 112a-b. |
Synthesis of product 114 involved tandem cyclization with diethyl oxalate from commercial compound 113. The Pd/C reduces compound 114 and yielded 115. Ester 7 was hydrolyzed with KOH in aqueous solution of isopropyl alcohol, producing acid 116 with a 50% yield. Subsequent to the synthesis of acid 8, amidation was conducted utilizing various substituted anilines (117) alongside coupling agents, especially 1,1′-carbonyldiimidazole (CDI) in tetrahydrofuran (THF), resulting in the production of eleven derivatives (118a-k) as depicted in Scheme 20. The recently acquired N-aryl-3,4-dihydro-2H-benzo[h]chromene-2-carboxamide derivatives (118a-k) has enhanced regulatory and anti-proliferative characteristics relative to KL-1156. Compound 118i demonstrated significant inhibition of LPS-induced NF-κB transcriptional efficiency and strong anti-proliferative effects on NCI-H23 lung cancer cells of lines43.
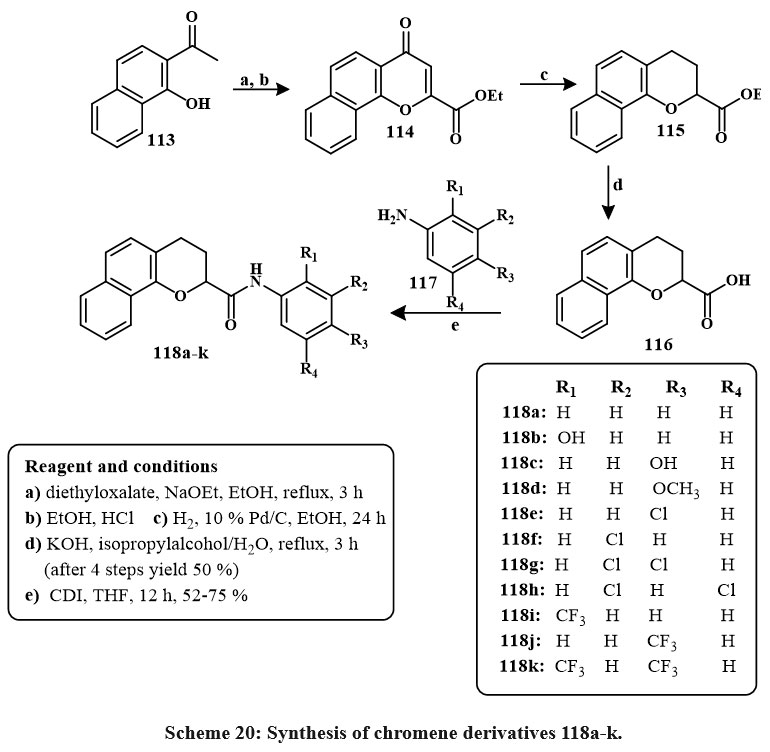 |
Scheme 20: Synthesis of chromene derivatives 118a-k. |
Under Sharpless conditions, the derivatives of chromene like O-propargylated 119 was interact with various azides conjugated, such as azides of alkyl or aryle, in THF to produce 120a, b, c, d, respectively, in high yield Scheme 21. All products were tested for cytotoxicity versus four lines in cancerous cells. It has been observed that in A549 lung cancerous cells, DU145 prostate cancerereous cells, HeLa cervical cancerous cells, and MCF7 breast cancerereous cells, compounds like of 120a, c, d, b exhibited potential activity at <20 lM concentration44.
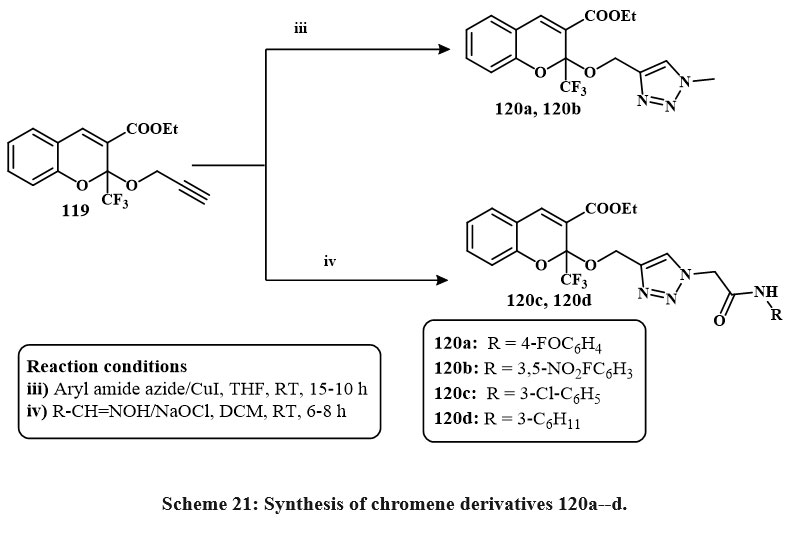 |
Scheme 21: Synthesis of chromene derivatives 120a–d. |
Product 122 was produced by refluxing compound 121 with triethyl orthoformate in acetic anhydride and benzene for 2 hrs. The subsequent reactions of 122 resulted in the synthesis of fused heterotetracyclic complexes with a pyrimidine nucleus at the positions -2, -3 and a 4H-chromene group. Exposure of 122 to NH3 gas in methanol at room temperature for one hour and obtained the formation of the open-chain product 123 (Scheme 22). The subsequent cyclization of 123 in ethanolic piperidine in the reflux yielded compound 124. Furthermore, the reaction of imidate 122 with methylamine in ethanol at ambient temperature for one hour yielded the open-chain product 125. The cycloaddition yield 126 was produced by reacting 122 with hydrazine hydrate. Compounds 126, 123, 124, and 125 exhibited better anticancer activity towards the MCF-7, HCT, and HepG-2(IC50 = 3.0-9.4 µM, 1.7-7.4 µM, and 3.0-6.2 µM, respectively) compared to Vinblastine and Colchicine, while compound 126 (IC50 = 1.7 µM) performed better than Doxorubicin45.
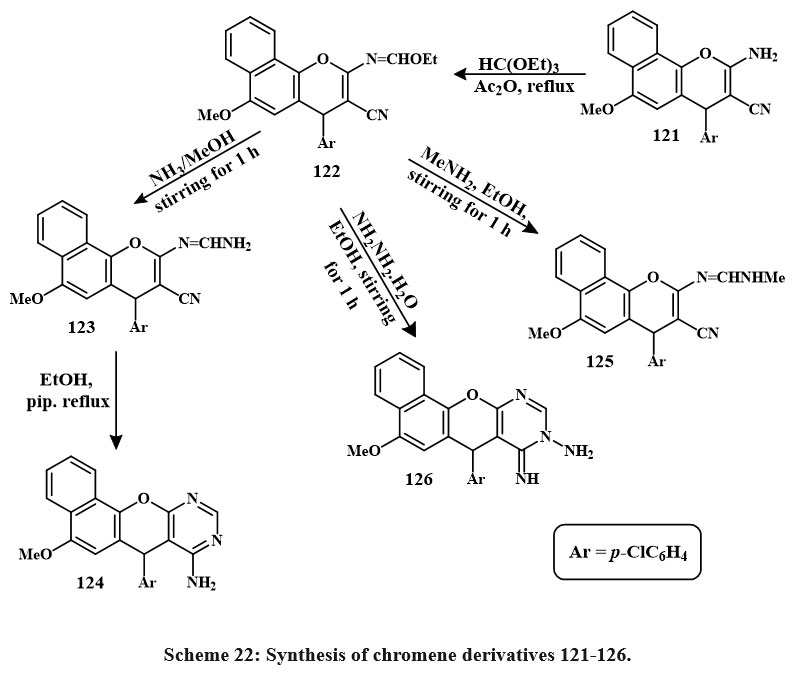 |
Scheme 22: Synthesis of chromene derivatives 121-126. |
Treatment of 4-chloro-1-naphthol with 127a-i formed the series of products 128a–i. Similarly, the chmical reaction of 4-chloro-1-naphthol with 129a-h yields 130a–h, as shown in Scheme 23. Compounds 130e, 128c, 130f, b, d, 128d, 130c, a were the prominent active compound towards the MCF-7 with IC50 amount of 2.4-6.0 µg/mL, 130a towards the HCT-116 with the IC50 amount of 2.7 µg/mL and 130a, 128e, a against HepG-2 (IC50 = 3.5-5.0 µg/mL) as compared to standard drug47, 48.
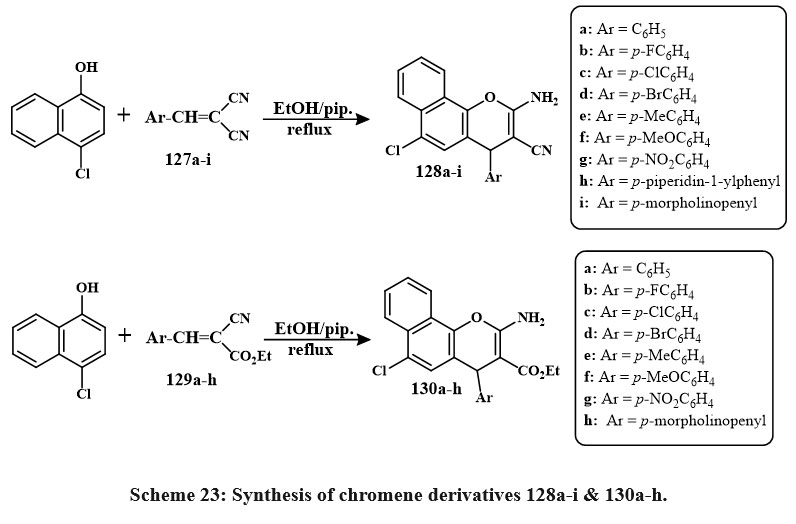 |
Scheme 23: Synthesis of chromene derivatives 128a-i & 130a-h. |
Scheme 24 outlines the series of preparation of a unique derivatives of chromene (135a-t) via the reaction of 2-hydroxy-benzaldehyde 131a-c with either acrolein 132a or 3-methylacrolein 133b in the presence of K2CO3 to produce chromene-3-carbaldehyde 134a-d. The chemical reaction, conducted under reflux in 1,4-dioxane, progressed via intermediates 133a-d. The resultant compounds 135a-t were assessed for anticancer efficacy towards several cancerous lines cell. The hydantoin derivative 135o, including a 6-bromo-2-methyl-2H-chromene core, had the most significant anticancer action, with efficacy comparable to that of cisplatin. The IC₅₀ values of 135o were established as 17.5 µM for A549 (epithelial adenocarcinoma), 10.6 µM for K562 (leukemia), 15.3 µM for MCF-7 (breast adenocarcinoma), 24.8 µM for MOLT-4 (acute lymphoblastic leukemia), and 25.2 µM for NIH/3T3 (mouse embryo fibroblasts, non-cancerous cells4).
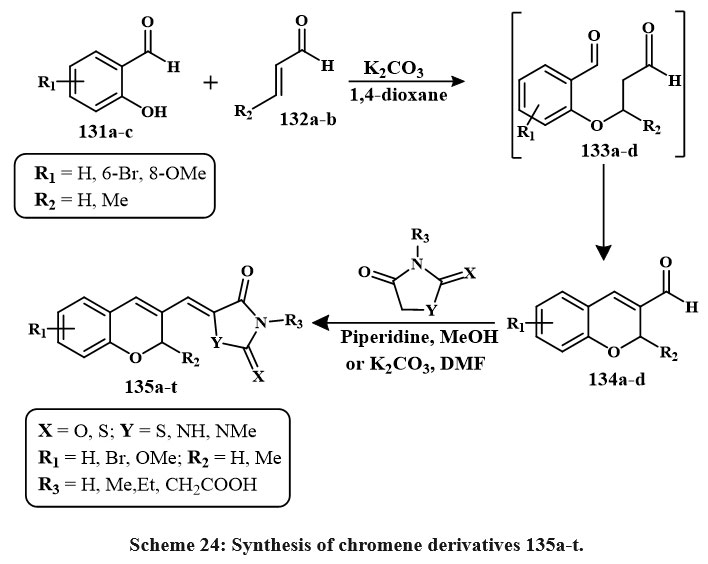 |
Scheme 24: Synthesis of chromene derivatives 135a-t. |
Compound 136 was synthesized according to the reported method; then, by a sequence of three-step synthesis, key intermediate 133a-d were prepared in Scheme 25. Then, 137b and 137d were used to synthesize the series of chromene derivatives that can be seen in Scheme 26. In vitro, cytotoxicity was assessed on MCF-7 human breast carcinoma cells. Compounds 137a, 137b, 138f, 140c, and 140f showed potential for breast tumor cell pharmacological inhibitors. The most effective compounds were 137a and 138f with an IC50 amount of 0.007 µM, which had nearly twise the efficiency of colchicine with an IC50 amount of 0.013 µM. There are mainly three compounds (137b, 140c, 140f) with an IC50 amount of 0.008 µM (Fig. 7) demonstrated approximately one half of the efficiency of the colchicine. Two compounds, like 138a and 138h with an IC50 amount of 0.011 µM, inhibited breast cancerous cells almost as well as the +favourable control of results with an IC50 amount of 0.013 µM. Most of the compound like 139d with IC50 vaules of 0.029 µM showed roughly one half of the action of the original which confirms the favourable control. Lastly, the compounds like of 138c and 138g with an IC50 amount of 0.033 µM, and 0.039 µM respectively are the lowest reactive48.
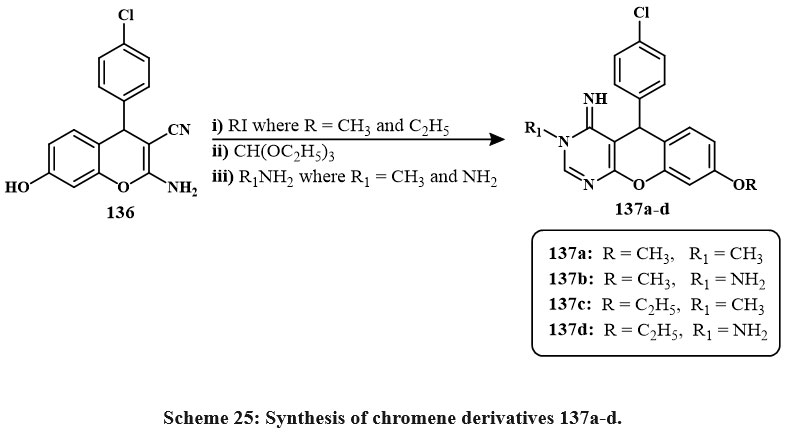 |
Scheme 25: Synthesis of chromene derivatives 137a-d. |
The initial compound, chromene derive compound like of 4-oxo-4H-chromene-3-carbonyl based chloride 141, was hydrolyzed to generate 4-Oxo-4H-1-benzopyran-3-carboxylic acid 142, and condensation with methyle alchol at ambient temperature yielded methyl 4-oxo-4H-1-benzopyran-3-carboxylate. Due to the varied reaaction ability of chromene derive compound like 4-oxo-4H-chromene-3-carbonyl based chloride 1 and its methyl ester 143 with elementry polyamines (a), two branches of materials were produced, only 144a and 145a showed in Scheme-27. The extremely violent melanoma lines cell A375 was used to test compounds 144a and 145a at both 50 and 75 µM concentrations in vitro. Compound 145a outperformed compound 144a in inducing apoptosis and cell cycle arrest49.
 |
Scheme 26: Route for the formation of chromene-based derivatives. |
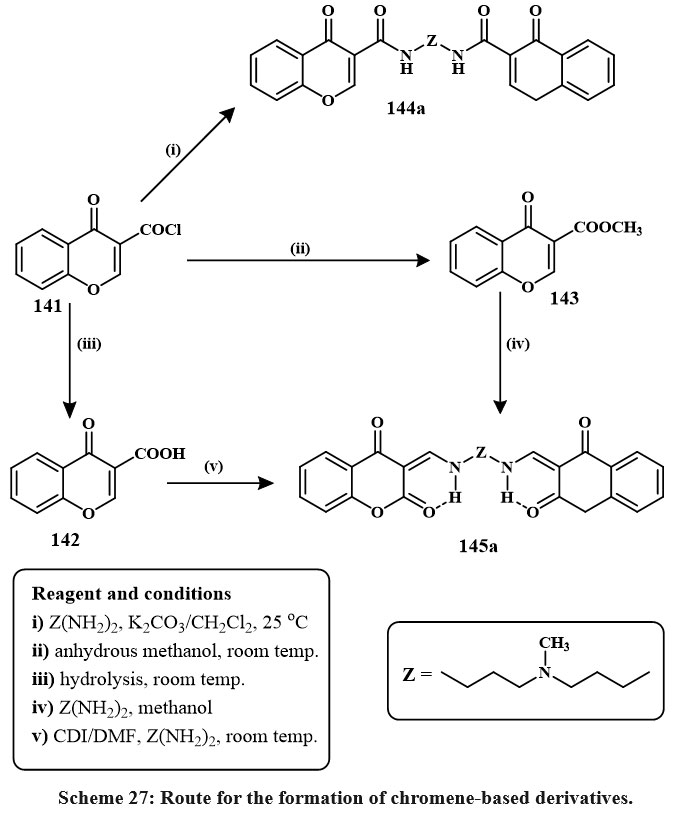 |
Scheme 27: Route for the formation of chromene-based derivatives. |
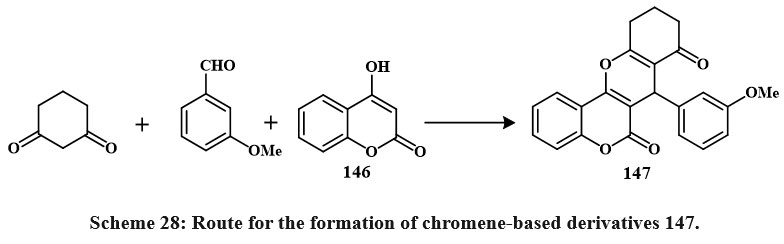 |
Scheme 28: Route for the formation of chromene-based derivatives 147. |
147 was obtained by synthesizing coumarin 146, cyclohexadione, and arylaldehydes in various solvents (ethanol, acetic acid, acetonitrile, and benzene) (Scheme 28). Boiling acetic acid produced the maximum product (66-78 %). Four cancerous cell lines were employed to check these compounds’ cytotoxic effects. The compound 147 demonstrated the highest activity on HeLa, LS180, MCF-7, and Raji (IC50 = 89.9, 99.7, 58.6, and 49.2 µM (Shafiee et al., 2011). Synthesis for compound 148 has been shown in Scheme 29. Excitingly, research on drug-resistant cancerous cells such as mitoxantrone- (HL-60/MX2) and camptothecin- (CCRF-CEM/C2) has shown that 148 can selectively kill these cells while leaving parental cancer cells alone. The mechanism of 148 selectivities toward drug-resistant cancer cells is being studied in depth because of its ability to limit tumor cell development by inducing apoptosis. These findings point to 148 as a potentially effective therapeutic target for multidrug-resistant malignancies50.
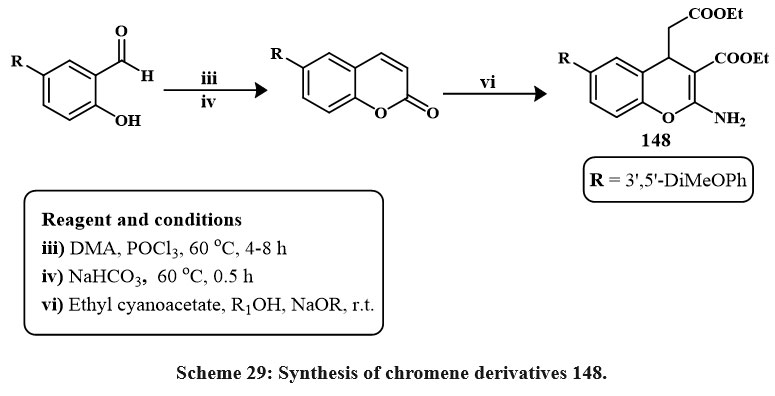 |
Scheme 29: Synthesis of chromene derivatives 148. |
One pot synthesis
The standard reaction condition was then applied to a wider range of substrates, including malononitrile, 5,5-dimethylcyclohexane-1,3-dione, and N-substituted indole-3-carbaldehydes 149a-j were synthesized, featuring electron-donating (OMe) and electron-loving (F, Br, and NO₂) groups at the 5th position of the ring, as depicted in Scheme 30. The derivatives 150a-j were synthesized with satisfactory yields, varying from 80% to 85%, from obtained using the newly established one-pot, 3-component chemical reaction with various conjugate indole-3-carbaldehydes. The new chromene based derivatives like indole-tethered 150a–j were tested for anticarcinoma activity towards lung carcinoma A549, the breast carcinoma MCF-7, and prostate carcinoma PC-3. As a conventional main drug, doxorubicin proved that most derivatives are cytotoxic. The compounds 150c and 150d, with a fluorine conjugate at the position of 5th place in the ring of indole, were the prominent effective versus all tested lines of cell, with IC50 values with range of 7.9 to 9.1 µM. Furthermore, materials 150g and 150h exhibited moderate inhibitory effects toward three cell lines, showing IC50 amount from 10.5 to 12.6 µM. Compounds 150e and 150f have lower potency in MCF-7 (IC50 values: 58.9-62.8 µM) and did not inhibit A549 or PC-3 cells51.
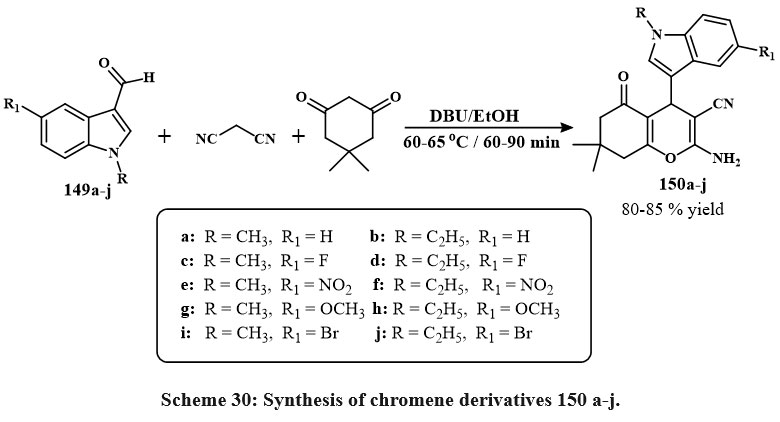 |
Scheme 30: Synthesis of chromene derivatives 150 a-j. |
Compound 151 was produced by one-pot, 3-component reactions of 6-chloro-3-hydroxychromone, aromatic aldehydes and ethyl cyanoacetate in ethanol with triethylamine as solvent and reflux Scheme 311. The compounds 151 were tested against the SW480 line cell and human umbilical vein endothelial cells (HUVEC), and it appears to be a potential candidate for a cytotoxic agent. 10.8 ± 1.5 and 57.2 ± 3.2 µM, respectively, were the IC50 values1.
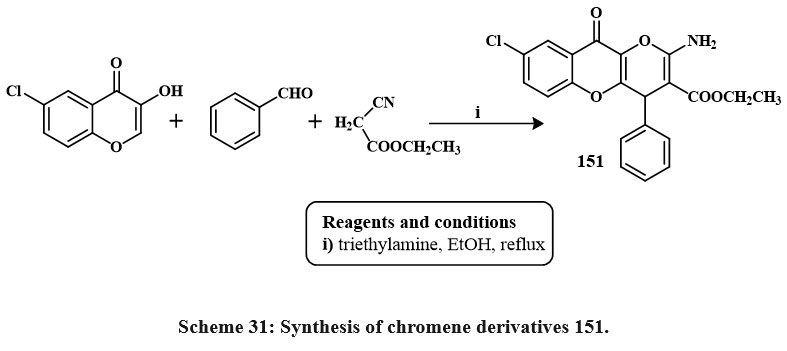 |
Scheme 31: Synthesis of chromene derivatives 151. |
A chemical reaction that involves three different reactants involving chromen based derivatives like of 3-hydroxy-4H-chromen-4-one, various carbon ring based aldehydes 152a,b,c, (Scheme 32) and either malononitrile 153a or ethyl cyanoacetate 153b is carried out in ethyle alcohol at reflux with the existance of catalytic quantities of nano particles MgO. Most of the obtained compounds demonstrated inhibitory properties towards the formation of a colorectal cancerous line cell (Caco-2). The compound with the lowest IC50 value was 37 mg/mL, identified as compound 154i, followed by compounds 154g and 154b. A significant portion of the synthesized compounds generally exhibited IC50 values below 100 mg/mL when tested on this specific cell line52.
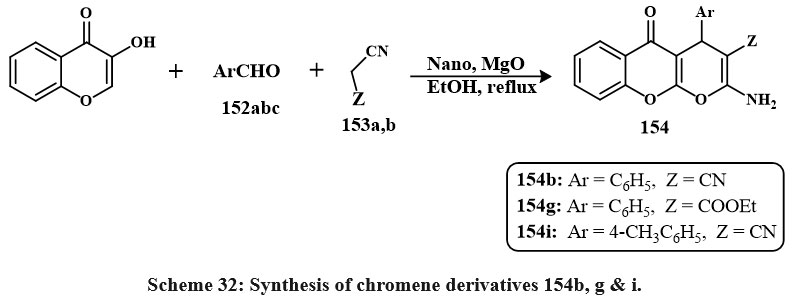 |
Scheme 32: Synthesis of chromene derivatives 154b, g & i. |
One-pot multicomponent reactions of malononitrile and dimedone with various azo aldehydes, involving Knoevenagel condensation and Michael addition, were conducted in ethanol employing DBU as a catalyst. The respective azo-chromene analogues were synthesized with satisfactory yields, as illustrated in Scheme 33. In MCF-7 cells, cytotoxicity was dose-dependent for compound 155, celecoxib, and anastrozole with IC50 values of 19.8, 26.2, 24.7 μg/ml, respectively. Comparing the IC50 of material 155 to those of anastrozole and celecoxib demonstrates that it possesses more cytotoxic behaviour2.
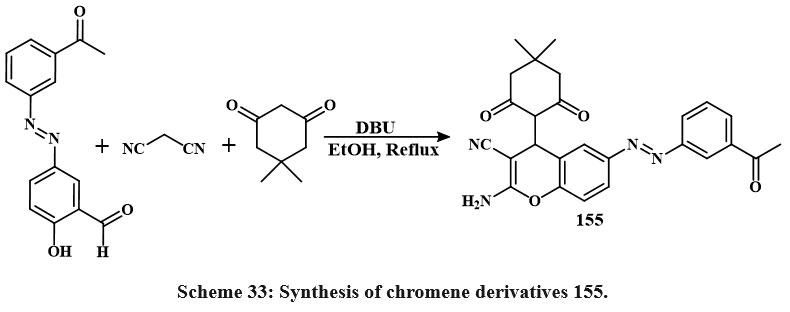 |
Scheme 33: Synthesis of chromene derivatives 155. |
The desired 4H-benzo[h]chromene derivatives, 157 and 158, were synthesized through a one-pot, 3-component condensation of 4-methoxy-1-naphthol, 4-methoxybenzaldehyde, and malononitrile in an ethanolic solution with piperidine, conducted for two minutes at a temperature of 140 ℃. After that, compounds 157 and 158 were utilized to synthesize several different chromene derivatives, and the chemical steps involved in their synthesis are outlined in Scheme 34 below. Compounds 161, 163, 160a, 169, and 170 (170, 169, 160a, 163 and 161) were the prominent derivatives versus MCF-7 cancerous cells, being 1.3, 1.5, 5.1, 5.6, 6.8, and 3.6, 4.3, 14.8, 1, 16, 19.7 time effective than colchicine and vinblastine, accordingly. Additionally, some compounds 159, 160b, 167, 166 and 168 were also active. Compounds 160a, 170, and 169 were also 2.2, 2.9, and 3.3 time effective than colchicine and vinblastine towards HCT-116 cancer cells. Meanwhile, compounds 159, 161, 167, 166, 160b, 163, 162, 165, 157, 168, and 158 showed good active towards HCT-116 cancerous lines cells. However, compounds 160a, b, 170, 169, and 159 were the most active in HepG-2 cytotoxicity (5.1, 1.2, 5.1, 5.8, 6.6 and 11.8, 2.7, 11.8, 13.3, 15.1, time effective than colchicine and vinblastine). Compounds 157, 162, 163, and 167 were 1.1, 1.3, 1.5, and 2.0 times high effective than colchicine against HepG-2 cancer cells5.
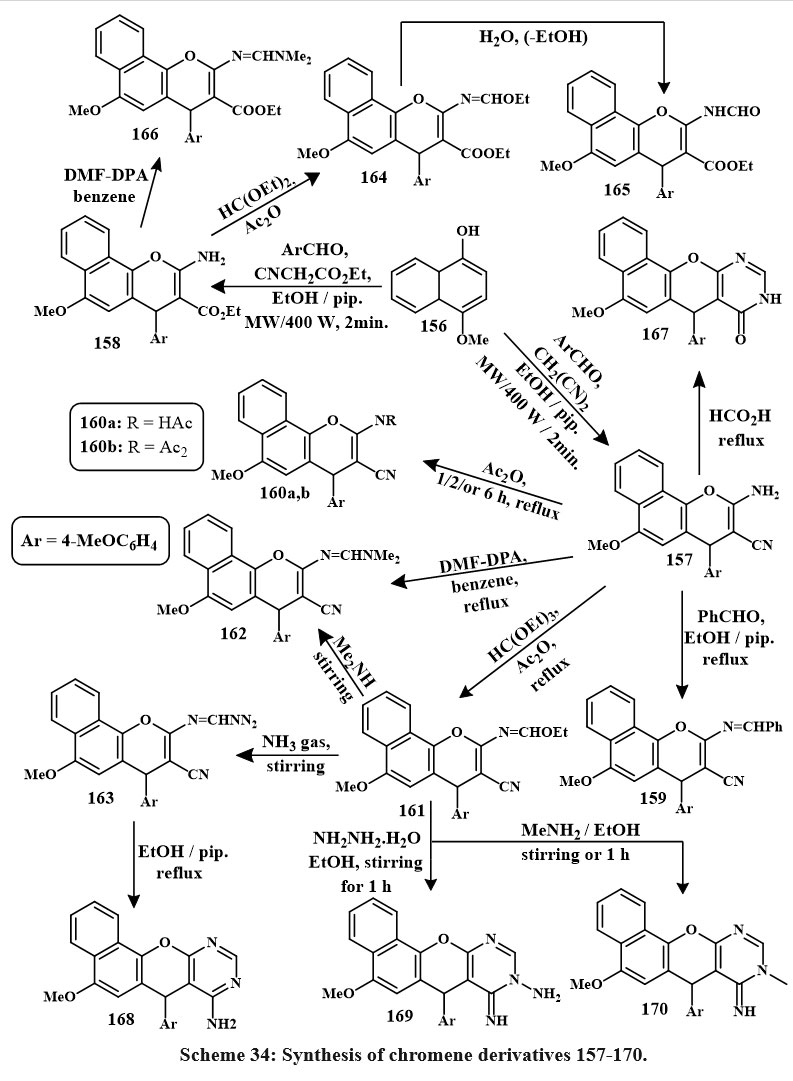 |
Scheme 34: Synthesis of chromene derivatives 157-170. |
Overall, piperidine has been incorporated into the resulting mixture of 3-dimethylaminophenol, substituted aryl-benzaldehyde, and malonitrilein in the form of an ethanol-based solvent (Scheme 35). The resulting solution was agitated for a 12-hour period at 35 ºC. Upon cooling, the accumulated material was filtered, cleaned with cold ethyle alcohol, and condensed from the solvent. All drugs were tested for Src kinase inhibition and cell growth in leukemia CCRF-CEM and HT-29 line cells. The 171c, 171h, 171i, and 171k conjugated chromenes with IC50 amount of 11.1 to 18.3 μM in Src kinase. Compound 171c selectively inhibits Src values of IC50 is of 11.1 μM, compared to other kinases, including EGFR (IC50 > 300 μM), Csk (101.7 μM), and Lck (46.8 μM). At 50 μM concentration, 3-chlorophenyl conjugated thiazole 171v and 2-chlorophenyl substituted 171u derivatives of chromene that reduced CCRF-CEM and HT-29 cell lines proliferation by 50% and 80%, respectively53.
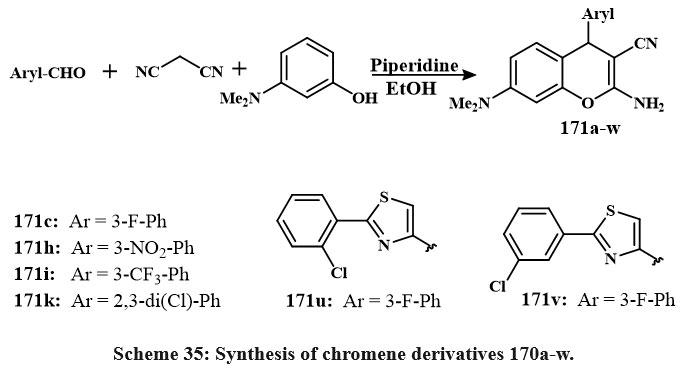 |
Scheme 35: Synthesis of chromene derivatives 170a-w. |
Microwave-assisted synthesis
Ethanolic solutions of 173a-h were produced by exposing naphthalene-2,7-diol, malononitrile, aromatic aldehydes 172a-h, and piperidine to microwave irradiation at 140°C for two minutes (Scheme 36). The highest yield of compound 173a-h was achieved at 400 W and 2 min. Compounds 173a-h showed the strongest efffective versus SKOV-3, PC-3, and HeLa, that exihibits the IC50 values of 0.9-3.1, 0.8-3.7, and 1.7-5.7 µM. Some compounds 173f, 173h, 173c, 173a, 173e, and 173b showed strong efficiency towards the MCF-7/ADR cells (8.6-11.5 µM IC50) and mild efficiency against HFL-1 and WI-38 cells (19.1-30.1 µM IC50). The compounds like of 173a, 173b, 173c, 173d, 173e, and 173f exihibits P-gp progression and good function in the MCF-7/ADR lines, with IC50 amount of 18.7-34.6 µM, outperforming than the Doxorubicin with IC50 values of 50.9 µM. Additionally, Rho123 aggregation experiments demonstrated that chemicals 173a, 173b and 173d suppressed P-glycoprotein accumulation and efflux activity, whereas compounds 173c and 173d promoted apoptosis and triggered G1 phase cell cycle stop, 173a and 173b at S phase, and 173e at G1/S phases54.
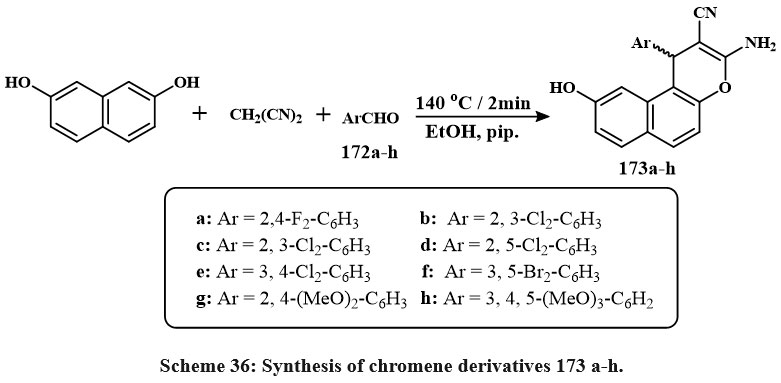 |
Scheme 36: Synthesis of chromene derivatives 173 a-h. |
Scheme 37 delineates the preparation of the required molecules 175a-d. The procedure entailed the reaction of 7-methoxy-2-naphthol with various aromatic aldehydes in microwave irradiation at 140°C for two minutes, resulting in compounds 175a-d. These products possess diverse conjugated (2,6-F₂; 2,3-Cl₂; 2,5-Cl₂; and 3,5-Br₂-2-OMe) affixed to the phenyl group of the 1H-benzo[f]chromene structure. Among the 1H-benzo[f]chromene-2-carbothioamide derivatives, the difluoro-substituted molecule had the greatest cytotoxic efficacy with IC50 amount of 5.4, 4.5 µM for MCF-7 and HepG-2, compared to the other halogenated derivatives tested versus PC-3, HepG-2, and MCF-7. The halogenated derivatives showed superior activity towards the PC-3 line cell with an IC50 amount of 1.1-2.7 µM) contras to the MCF-7 and HepG-2 cell lines55.
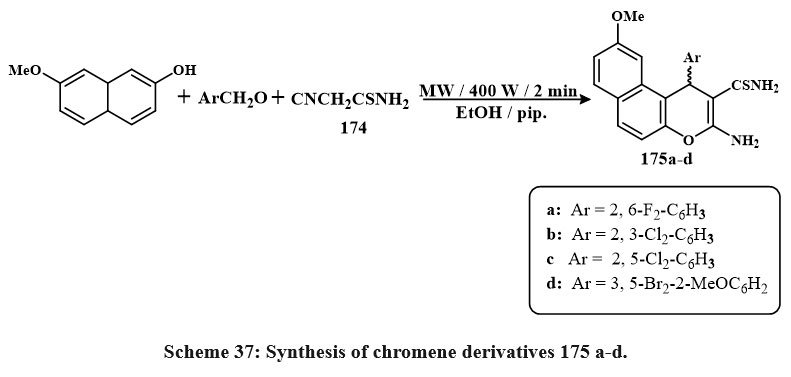 |
Scheme 37: Synthesis of chromene derivatives 175 a-d. |
The compounds (177e, f, m) illustrated in Scheme 38 were obtained by the chemical reaction of chromen-2-one with a variety of aryl aldehydes 176e, f, m, piperidine 4 and malononitrile 3 in ethanol in microwave irradiation for 2 minutes. All synthesized compounds were tested through antitumor agent testing using MCF-7, HepG-2, and HCT-116, human cancerous lines cell. Compared to doxorubicin, the standard cytotoxic drugs, compounds 177e, 177f, and 177m, were the very effective versus all tested cancerous lines cell. The IC50 values were found to be 2.7, 0.6, and 0.32 M for MCF-7, 3.1, 0.2, and 1.7 M for HCT-116, and 2.2, 0.5, and 0.4 M for HepG-2, respectively6.
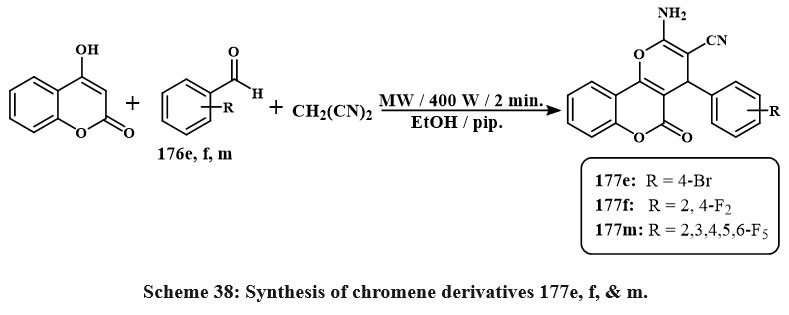 |
Scheme 38: Synthesis of chromene derivatives 177e, f, & m. |
Scheme 39 illustrates the chemical reaction of β-enaminonitrile 178 with nucleophilic agent such as acetic anhydride and triethyl orthoformate, leading to the formation of the 4H-benzo[h]chromene derivative 179. The main compounds in step 4 are useful starting materials for making a wide variety of new heterocyclic derivatives from 4H-benzo[h]chromene. Therefore, pyrimidine derivative 180 was produced when molecule 178 was condensed with formic acid. The cycloaddition product of aminoiminopyrimidine derivatives 181, respectively, was obtained after treating the imidate 179 with the hydrazine hydrate in a methanolic solution at room temperature for 1 hour. The aminoimino derivative 181 showed strong anticancer effective versus MCF-7, HCT-116, and HepG-2 cancerous cell lines, with IC50 amount of 0.45, 1.7, and 0.7 µg/mL. Compounds 179, 180, and 181 were chosen for cell cycle studies, apoptosis assays, caspase 3/7 activity assessment, and DNA fragmentation investigations. These 3-compounds of cytotoxic cause apoptosis by arresting the S and G2/M cell cycle. These chemicals also greatly reduce cancer cell invasion and migration56.
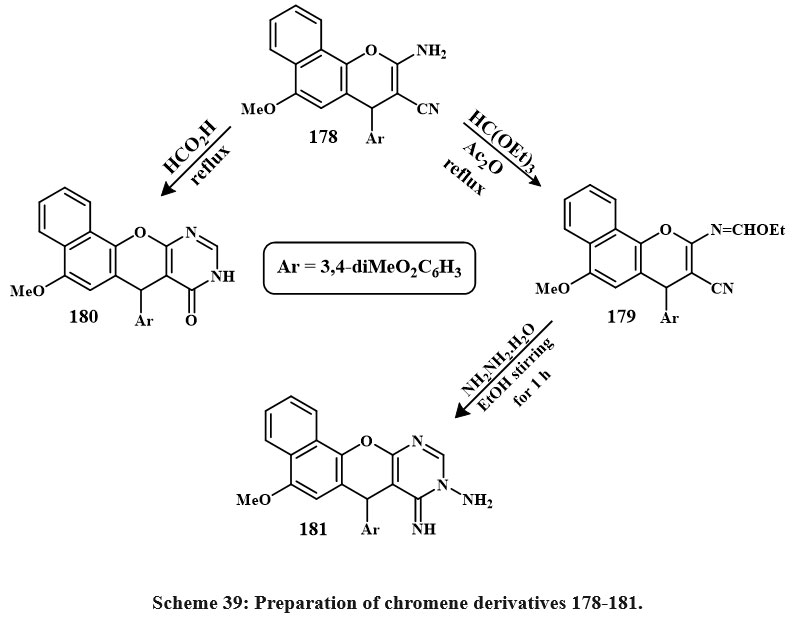 |
Scheme 39: Preparation of chromene derivatives 178-181. |
Scheme 40 depicts the production of 3-amino-1-aryl-1H-benzo[f]chromene-2-carbonitriles 183a–h. The chemicalreaction commenced by amalgamating 2-naphthol with aromatic aldehydes 182a–h and malononitrile in the solution of ethanolic piperidine, thereafter subjected to microwave irradiation at 140°C for two minutes. In a similar manner, as illustrated in Scheme 40, the reaction of 7-methoxy-2-naphthol compound with the identical array of aromatic aldehydes 182a–h and malononitrile yielded 3-amino-1-aryl-9-methoxy-1H-benzo[f]chromene-2-carbonitriles 184a–h57.
The synthesized compounds demonstrated strong and selective cytotoxic impact towards the three cancerous lines cell: MCF-7, HCT-116, and HepG-2. Compounds 183b-g, and 184b-e, and h exhibited the greatest activity against MCF-7 cells, with IC₅₀ values between 0.18 and 5.7 µg/mL. Compounds 183b-d, and g and 184b-e, and h exhibited increased efficacy against HCT-116 cells, with IC₅₀ amount of 0.6 to 2.5 µg/mL. Moreover, compounds 183b-d, f and g and 184b-e shown enhanced cytotoxicity (IC₅₀ = 0.6–3.9 µg/mL) relative to Vinblastine and Colchicine (IC₅₀ = 4.6 and 10.6 µg/mL, respectively). SAR research explained that the incorporation of halogen substituents (electron-loving groups) at the 4-position of the phenyl ring on the 1-position of the 1H-benzo[f]chromene core markedly improved antitumor activity. Conversely, electron-donating groups like of methyl or methoxy exhibited diminished efficacy. Moreover, the existance of a hydrogen atom at the 9-position was more advantageous for cytotoxic activity than a methoxy group57.
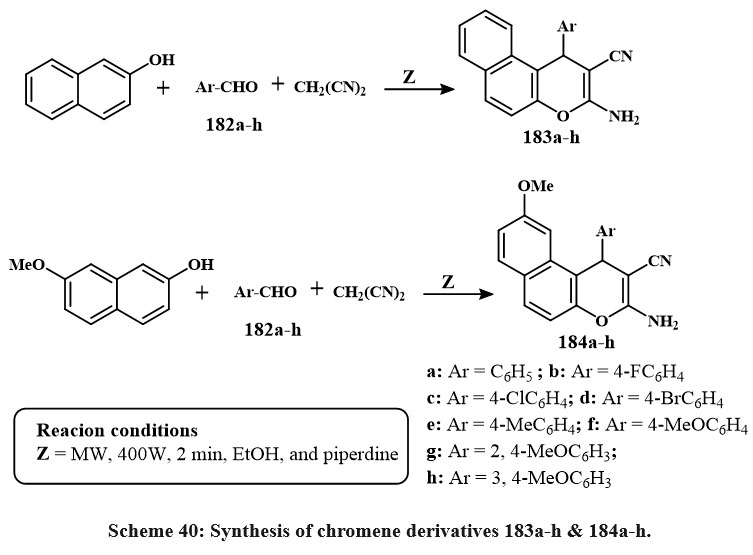 |
Scheme 40: Synthesis of chromene derivatives 183a-h & 184a-h. |
Other synthesized anti-cancerous chromene derivatives
There have been many additional chromene compounds created several of which have potent anticancer properties. At doses in the low range like of μM and nM, they have repeatedly been shown to have cytotoxic impact on cancerous-based cell lines.
Coumarin derivatives 185 Fig. 7, with the 4,5-dihydropyrazole moiety, may suppress telomerase. Compound 185 (R₁ = H, R₂ = COMe, R₃ = Ph) shown notable efficacy against the human gastric cancerous cell line SGC-7901, with an IC₅₀ amount of 2.69 µg/µL. Additionally, both this drug and its derivative 81 (R₁ = Br, R₂ = COMe, R₃ = Ph) demonstrated significant telomerase inhibition, with IC₅₀ amount of 2 µM and 1.8 µM, respectively58. Additionally, compound 185 (R1 = H, R2 = C2OH2N(nPr)2, R3 = 4-CF3C6H4) exhibited strong action with an IC50 amount of 0.98 µM. The in vivo study showed that this drug might suppress S180 and HepG2 tumor development and dramatically boost EAC tumor survival59. Compound 185, with R1 = H, R2 = C2OH2S(CH2)4CH3, R3 = 4-CF3C6H4, inhibited the telomerase with an IC50 of 0.92 µM60.
Chromene derivatives labelled as 186 Fig. 7, featuring an amine group at the 4 positions, were subjected to tests to assess their capacity for hindering impeding cell growth in HeLa cells and DNA preparation in Ehrlich cells61. This investigation revealed that prominent of the compounds has an activity solely in the one of such evaluations, and there was no discernible correlation between the outcomes of the two tests. Generally, many compounds were recognized as agents with antiproliferative and cytotoxic properties, displaying notably low amount of IC50 with ranging from 1.74-12.9 µM. The efficacy of these compounds was influenced through amine group and the conjugated located on the organic ring of the derivatives of chromene structure. Notably, the cyclo alkylamino groups and the phenylamino groups emerged as the very potent amino substitutions. Furthermore, the existance of an extra aromatic ring within the chromene structure, resulting in a tricyclic core, improved the biological efficacy.
Xing et al., studied 4H-chromene 187 Fig. 7 and chromene based derivatives’ cytotoxicity for cancerous cell lines in recent years. They have concentrated on identifying compounds that specifically attack drug-resistant cancerous cells to address this issue. A compound 187, with all groups are same (R1 = R2 = Et and R3 = H), was checked on cancerous cells with increased Bcl-2 group protein expression. It overcomes medication resistance by acting as an antagonist and is selectively cytotoxic to malignant cells62, 63. This compound elicited cell death via the endoplasmic reticulum (ER) pathway by enhancing ER stress and initiating the emission of ER-associated factors Ca2+. This substance also inhibited the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase. Compound 187, with R₁ = R₂ = Et and R₃ = 3,5-OMe, demonstrated μM cytotoxicity against over 11 cancerous cell lines, encompassing both hematologic and solid tumors cells50,64. This chromene was assessed in camptothecin- and mitoxantrone-resistant cancerous line cell (CCRF-CEM/C2 and HL-60/MX2). Remarkably, it normally eliminated drug-resistant cells over non-resistant ones. Analogues of compound 187 were generated by analyzing its structure and activity. According to Mooring et al., study, these analogues have the same conjugated ways are present in R3, though with R1 = R2 = CH2C≡CH65. One compound showed increased effectivity in the multi-resistant drug HL60/MX2 line cells, with an average values IC50 of 640 nM.
Nonsteroidal derivatives of the chromene 188a and 188b Fig. 7 identified particular inhibiting agents of endothelial cells proliferation through an assessment of primary human vein endothelial cells66. These compounds share structural similarities with Alternariol natural derivatives, which also display anti-cancer effects.
Compound 189 Fig. 7 blocks heat shock protein 90 and suppresses HIF-α client protein, making them antiproliferative agents. These chromenes showed nM activity in H1299 large cancerous lung cells. The dose-dependent antiangiogenic actions were tested in zebrafish embryos67.
Derivatives of neo-tanshinlactone 190, Fig. 7 were synthesized and evaluated for their anticancer efficacy across many different cell lines, which includes breast cancer (HS 587-T, MCF-7, SK-BR-3, MDA-MB-231, ZR-75-1), non-small cell cancers like of the prostate (LN-CaP, PC-3), lungs (A549), colon (SW620), nasopharyngeal (KB), skin (A431), and a vincristine-resistant KB subline (KB-VIN)68. Of these, chromene 190, which possesses a methyl group at R1 and either a methyl or ethyl group at R3, had the highest potency in cell growth inhibition and selectivity. The results indicate that the furan moiety and the configuration of aromatic ring substitution are essential in influencing anticancer activity. These compound displayed an ED50 values of 0.18 µg/mL for ZR-75-1 and 0.45 µg/mL for MCF-7. In another study, the same group examined 190 analogues by various ring position substitutions69. Once again, chromenes were found to be effective anti-cancer drugs with values of ED50 below the 1 µg/mL. Additionally, the highest outcomes again came from the groups of methyl and alkyl in R1 and R3 respectively.
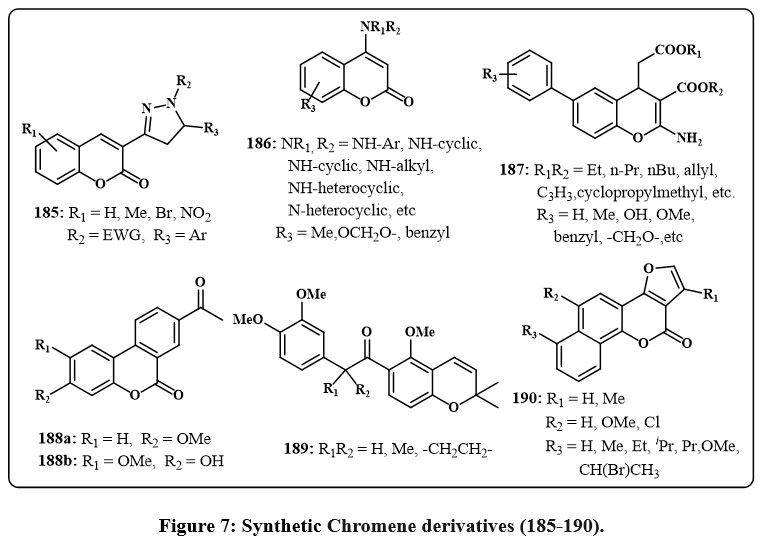 |
Figure 7: Synthetic Chromene derivatives (185-190). |
The anticancer efficacy of 2H-chromene 191 (Fig. 8) was assessed in various human cancer cell lines, including A549, K562, MCF-7, and MOLT-44. Chromene 191, characterized by a bromine atom at 6-position of the aromatic ring and a hydantoin moiety (X = O, Y = NH, and R2 = H) at the 3-position, shown considerable cytotoxic action, with IC50 values akin to those of cisplatin. Furthermore, compounds featuring a 6-membered ring and a 4-methoxyphenyl group in the amine moiety (R3 = H or Me) exhibited significant anticancer activity towards many cancerous lines of cell, with ED50 values between 0.32 and 0.008 µM. Chandra et al. examined the impact of compound 192 (Fig. 8) on estradiol-induced uterine hyperplasia in rate70. It is widely familiar with the name as estrogen that induced uterine tissue alternation can cause atypical hyperplasia and endometrial cancer. Compound 192 was delivered at dosages of 100 and 200 µg/kg, indicating its potential function in this investigation. These chromene chemical compounds triggered intrinsic apoptosis promotion of apoptosis by the inhibition of Akt function and the reduction of referred to as inhibitor of apoptosis protein levels. In addition, it prevented estradiol-induced uterine hyperplasia in rats.
Griffin and colleagues designed a set of inhibitors targeting protein kinase that are based on DNA (DNA-PK) to amplify the effectiveness for the faulty of DNA treatments for cancer therapy71. Chromenes labelled as 193 Fig. 8, featuring heterocyclic amines (piperidine) at the position-4, were investigated. Levels of inhibiting, quantified by IC50 values, ranged from 1.75 to 11.31 µM. The most potent result of IC50 was calculated of 1.75 µM that exihibits a morpholine group at 4-position in the chromene and additionally a group of methoxyl present at C6 of the ring. Compounds that impede endothelium cell growth are crucial for addressing conditions linked to angiogenesis, including solid tumors. The hypoxia-inducible factor (HIF) route is essential for cancerous cells’ adjustment to hypoxic environments, facilitating therapeutic resistance and establishing it as a significant target for cancer therapy. In this context, 2H-chromenes 194 Fig. 8 exhibited the capacity to suppress HIF-1 in the hypoxia-treated people glioma cell line LN229-HRE-Lux65, 72.
Blagg et al., Bras and colleagues investigated novobiocin analogues 195 and 196 Fig. 8, for their potential as antiproliferative agents and Hsp90 inhibitors, an attractive target in anticancer drug development73-76. Blagg and colleagues showed that the derivative of chromene 195, with methyl groups at R2 and R1 at the aromatic ring’s 8 position, inhibited Hsp90 protein-folding. Chromenes 195, including conjugate of 2-indolyl moieties, was tested with antiproliferative properties in multiple cancerous lines cell, including colon (HCT116), breast (SkBr3, MCF-7), and pancreatic (PL45) cancer. This exploration yielded numerous intriguing compounds with low IC50 values. Bras and colleagues examined derivatives 196 without the noviose moiety. Positions 4 and 7 needed tosyl moiety substitutes for optimal results. These compounds killed MCF-7 cells and degraded Hsp90 client proteins75. Coumermycin A1 derivatives 197 Fig. 8 inhibited Hsp90 protein folding77.
Synthesis and in vitro testing of coumarins 198 Fig. 9, with the hydrazide-hydrazone constituent was evaluated for its antiproliferative efficacy towards the human drug-resistant pancreatic cancer (Panc-1) and drug-sensitive cell lines (Hep-G2 and CCRF). Compound 198 that containing R1 = 6-Br and R2 = 2-thiophene cytotoxically active towards all examined cells (IC50: 3.60-6.50 mM) and caused apoptotic and G2/M gene expression in resistant Panc-1 cells78. Synthetic 4-Arylcoumarin numbered 199 Fig. 9, evaluated for their ability to combat cancer. Compound displayed the most promising outcomes by effectively restraining the progress of the cell lines of CEM leukemia even at very low concentrations like of 0.083 and 0.52 µM. This particular compound hindered tubulin assembly to induce apoptosis and prompted cell death through an alternative pathway that doesn’t involve apoptosis79.
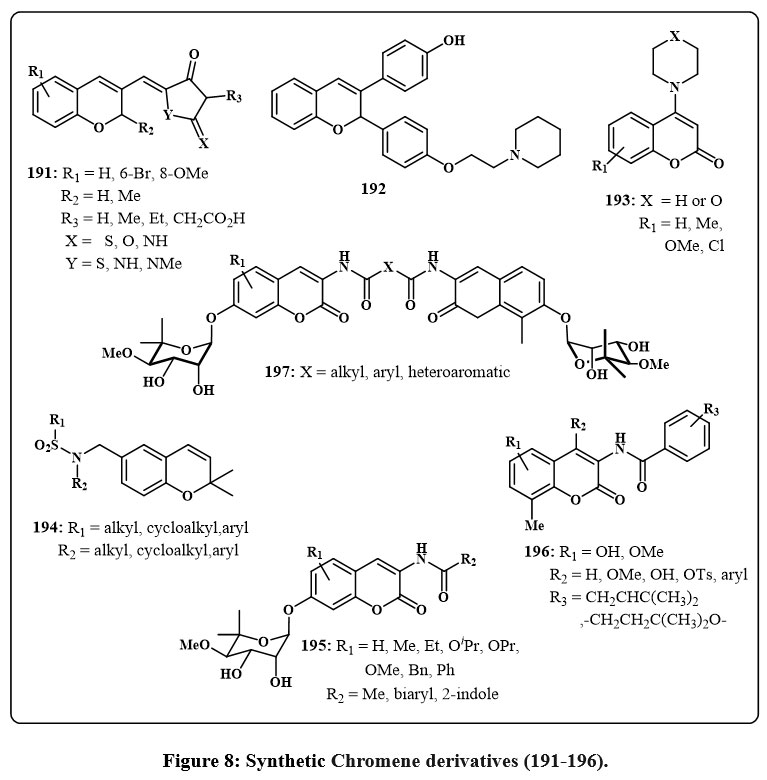 |
Figure 8: Synthetic Chromene derivatives (191-196). |
Analogous chromenes 200 Fig. 9, with an amine component in the 4 positions rather than of group of furan ring, inhibited tumor cell development80. The evaluation encompassed breast cancerous lines cell (SK-BR-3, MDA-MB-231, ZR-75-1), large cancerous cell of lungs (A549), prostate (DU145), along with cell lines of KB and KB-VIN. The amine component included conjugate group of aromatic, aliphatic, and heterocyclic, with the aromatic conjugate arrangement exhibiting the most potency, producing ED50 amount from 0.01 to 5.8 µM across the evaluated cell lines, making it the most convenient. Additionally, the 3-methylphenyl and 4-methoxy moieties with R1 = H showed the highest ED50 values ranging from 0.01-0.76 µM), indicating cell growth inhibition. The same authors generated similar chromenes 201 Fig.981. Certain compounds had a 5- or 6-membered ring linked to the aromatic chromene moiety rather than an additional chromene structure of 200 and also explored for the amine substituent.
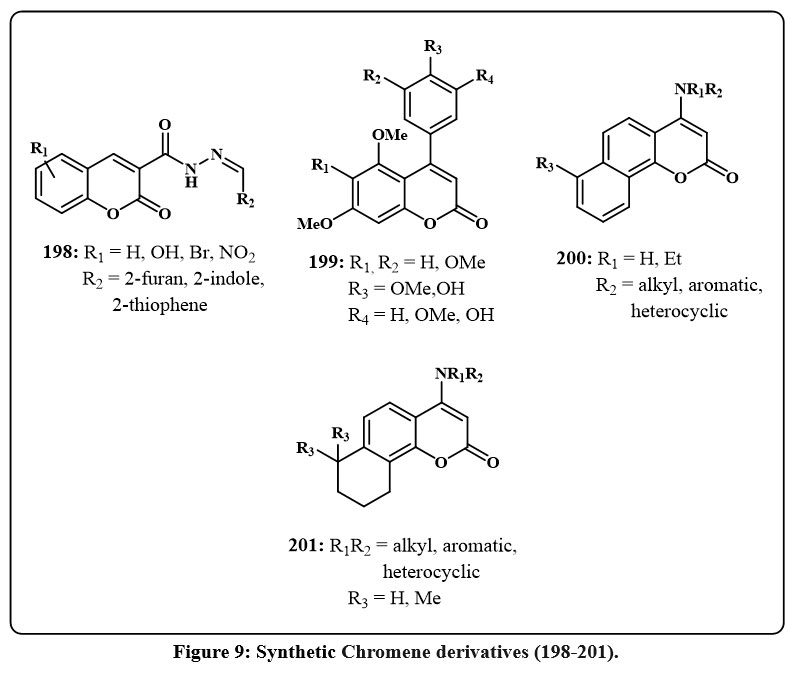 |
Figure 9: Synthetic Chromene derivatives (198-201). |
Conclusion
This review gives a current perspective that highlights the significance of the chromene structure in bioactive compounds. The structure of chromene is present in numerous natural compounds and has demonstrated interactions with diverse biological objectives, frequently providing a significant therapeutic advantage. The ring structure of 2H/4H-chromene has been utilized as the basis for creating new frameworks that show promise for identifying anticancer drugs and other biological uses. Microwave irradiation technique, one-pot preparation, and metal-catalyzed procedures, and other ecologically friendly methods are all available for creating the 2H/4H-chromene ring and derivatives of chromene. Among these, condensation and addition reactions like Michael addition and aldol condensation preparation methods are normally used to produce 4H-chromene analogues due to the ease with which starting materials can be obtained and the ability to yield a larger quantity of the final product via one-pot multicomponent preparation. The insertion of a wide range of C3-ester substitutions and C4-benzopyran alterations, particularly those involving changed aryl functionalities on chromene, is more beneficial for boosting anticancer activity. This current review describes various design approaches for 2H/4H-chromene compounds. These results highlight structural qualities that medicinal chemists can use as a reference when developing powerful chromene compounds.
Future Outcomes
The domain of chromene chemistry provides motivation for medicinal chemists and biopharmaceutical organizations. The existing body of knowledge represents just a fraction of the significant possibilities within this particular chemical group. In their quest to find novel chemicals with increased biological efficiency, committed researchers still have many unexplored areas to discover.
Acknowledgement
The authors would like to thank Jazan University for the valuable support.
Funding Sources
This article received no external funding.
Conflict of Interest
The authors declare that there are no conflict of interest regarding the publication of this paper.
References
- Sabouri, S., Faghih-Mirzaei, E., & Abaszadeh, M. Acta Chim. Slov., 2022, 69(4), 920–929. https://doi.org/10.17344/acsi.2022.7754.
CrossRef - Bhuvaneswari, K., Sivaguru, P., & Lalitha, A. J. Chin. Chem. Soc., 2020, 67(10), 1877–1886. https://doi.org/10.1002/jccs.201900481.
CrossRef - Okasha, R. M., Alblewi, F. F., Afifi, T. H., Naqvi, A., Fouda, A. M., Al-Dies, A. A. M., El-Agrody, A. M., Belmont, P., & Bunce, R. A. Molecules, 2017, 22(3). https://doi.org/10.3390/molecules22030479.
CrossRef - Azizmohammadi, M., Khoobi, M., Ramazani, A., Emami, S., Zarrin, A., Firuzi, O., Miri, R., & Shafiee, A. Eur. J. Med. Chem., 2013, 59, 15–22. https://doi.org/10.1016/j.ejmech.2012.10.044.
CrossRef - Sawyers, C. Nat., 2004, 432(7015), 294–297. https://doi.org/10.1038/nature03095.
CrossRef - El-Agrody, A. M., Fouda, A. M., Assiri, M. A., Mora, A., Ali, T. E., Alam, M. M., & Alfaifi, M. Y. Med. Chem. Res., 2020, 29(4), 617–629. https://doi.org/10.1007/s00044-019-02494-3.
CrossRef - Al-Dies, A. M., Abd El-Wahab, A. H. F., Alamri, A. A., Borik, R. M. A., Mohamed, H. M., Assirey, E. A., Alsehli, M. H., Moussa, Z., Alzamly, A., Mehany, A. B. M., Elhenawy, A. A., El-Agrody, A. M. J. Molec. Struc., 2025, 1322(2), 140289. https://doi.org/10.1016/j.molstruc.2024.140289.
CrossRef - Patil, S. A., Patil, R., Pfeffer, L. M., & Miller, D. D. Fut. Med. Chem., 2013, 5(14), 1647–1660. https://doi.org/10.4155/fmc.13.126.
CrossRef - Fayed, E. A., Bayoumi, A. H., Saleh, A. S., Ezz Al-Arab, E. M., & Ammar, Y. A. Bioorg. Chem., 2021, 109, 104742. https://doi.org/10.1016/j.bioorg.2021.104742.
CrossRef - Fouad, S. A., Hessein, S. A., Abbas, S. Y., Farrag, A. M., & Ammar, Y. A. Croa. Chem. Acta, 2018, 91(1), 99–107. https://doi.org/10.5562/cca3266.
CrossRef - Iwashima, M., Mori, J., Ting, X., Matsunaga, T., Hayashi, K., Shinoda, D., Saito, H., Sankawa, U., & Hayashi, T. Biol. and Pharma. Bull., 2005, 28(2), 374–377. https://doi.org/10.1248/bpb.28.374.
CrossRef - Kiem, P. Van, Nhiem, N. X., Anh, N. H., Yen, D. T. H., Cuong, N. T., Tai, B. H., Yen, P. H., Nam, N. H., Minh, C. Van, Chinh, P. T., Jeon, Y. H., Park, S. J., Kim, S. H., & Kwon, S. H. Bioorg. Chem., 2020, 104, 104268. https://doi.org/10.1016/j.bioorg.2020.104268.
CrossRef - Costa, M., Dias, T. A., Brito, A., & Proença, F. Euro. J. Med. Chem., 2016, 123, 487–507. https://doi.org/10.1016/j.ejmech.2016.07.057.
CrossRef - Mar, W., Je, K. H., & Seo, E. K. Arch. Pharm. Res., 2001, 24(3), 211–213. https://doi.org/10.1007/BF02978259.
CrossRef - Yang, Y. M., Hyun, J. W., Sung, M. S., Chung, H. S., Kim, B. K., Paik, W. H., Kang, S. S., & Park, J. G. Plan. Med., 1996, 62(4), 353–354. https://doi.org/10.1055/s-2006-957901.
CrossRef - Dong, Y., Morris-Natschke, S. L., & Lee, K. H. Nat. Prod. Rep., 2011, 28(3), 529–542. https://doi.org/10.1039/c0np00035c.
CrossRef - Wang, X., Bastow, K. F., Sun, C. M., Lin, Y. L., Yu, H. J., Don, M. J., Wu, T. S., Nakamura, S., & Lee, K. H. J. Med. Chem., 2004, 47(23), 5816–5819. https://doi.org/10.1021/jm040112r.
CrossRef - Aiello, A., Fattorusso, E., Luciano, P., Macho, A., Menna, M., & Muñoz, E. J. Med. Chem., 2005, 48(9), 3410–3416. https://doi.org/10.1021/jm0489915.
CrossRef - Du, L., Mahdi, F., Jekabsons, M. B., Nagle, D. G., & Zhou, Y. D. J. Nat. Prod., 2011, 74(2), 240–248. https://doi.org/10.1021/np100762s.
CrossRef - Guilet, D., Séraphin, D., Rondeau, D., Richomme, P., & Bruneton, J. Phytochemistry, 2001, 58(4), 571–575. https://doi.org/10.1016/S0031-9422(01)00285-0.
CrossRef - Aly, A. H., Edrada-Ebel, R. A., Indriani, I. D., Wray, V., Müller, W. E. G., Totzke, F., Zirrgiebel, U., Schächtele, C., Kubbutat, M. H. G., Lin, W. H., Proksch, P., & Ebel, R. J. Nat. Prod., 2008, 71(6), 972–980. https://doi.org/10.1021/np070447m.
CrossRef - Kharwar, R. N., Mishra, A., Gond, S. K., Stierle, A., & Stierle, D. Nat. Prod. Rep., 2011, 28(7), 1208–1228. https://doi.org/10.1039/c1np00008j.
CrossRef - Scio, E., Ribeiro, A., Alves, T. M. A., Romanha, A. J., Shin, Y. G., Cordell, G. A., & Zani, C. L. J. Nat. Prod., 2003, 66(5), 634–637. https://doi.org/10.1021/np020597r.
CrossRef - Ĺopez-Pérez, J. L., Olmedo, D. A., Del Olmo, E., Vásquez, Y., Solís, P. N., Gupta, M. P., & San Feliciano, A. J. Nat. Prod., 2005, 68(3), 369–373. https://doi.org/10.1021/np049642g.
CrossRef - Ma, J., Jones, S. H., & Hecht, S. M. J. Nat. Prod., 2004, 67(9), 1614–1616. https://doi.org/10.1021/np040129c.
CrossRef - Phan, V. K., Nguyen, H. D., Ha, V. B., Hoang, T. H., Chau, V. M., Le, M. H., Jung, J. L., & Young, H. K. Arch. Pharm. Res., 2005, 28(10), 1131–1134. https://doi.org/10.1007/bf02972974.
CrossRef - Alsharif, M. A., Khan, D., Ahmed, N., Mukhtar, S., Khan, P., Hassan, M. I., Almalki, A. S. A., & Obaid, R. J. Chem.Select, 2020, 5(2), 498–505. https://doi.org/10.1002/slct.201904096
CrossRef - Abdelatef, S. A., El-Saadi, M. T., Amin, N. H., Abdelazeem, A. H., Omar, H. A., & Abdellatif, K. R. A. Eur. J. Med. Chem., 2018, 150, 567–578. https://doi.org/10.1016/j.ejmech.2018.03.001.
CrossRef - Tan, S., He, F., Kong, T., Wu, J., & Liu, Z. Bioorg. and Med. Chem., 2017, 25(15), 4123–4132. https://doi.org/10.1016/j.bmc.2017.05.062.
CrossRef - El-Agrody, A. M., Fouda, A. M., & Khattab, E. S. A. E. H. Med. Chem. Res., 2017, 26(4), 691–700. https://doi.org/10.1007/s00044-016-1773-x.
CrossRef - Abd El-Wahab, A. H. F., Borik, R. M., Al-Dies, A. M., Fouda, A. M., Mohamed, H. M., El-Eisawy, R. A., Sharaf, M. H., Alzahrani, A. Y. A., Elhenawy, A. A., El-Agrody, A. M. Sci. Rep., 2024, 14(1), 9862. https://doi.org/10.1038/s41598-024-59193-2.
CrossRef - Halawa, A. H., Elaasser, M. M., El Kerdawy, A. M., Abd El-Hady, A. M. A. I., Emam, H. A., & El-Agrody, A. M. Med. Chem. Res., 2017, 26(10), 2624–2638. https://doi.org/10.1007/s00044-017-1961-3.
CrossRef - Widelski J, Popova M, Graikou K, Glowniak K, Chinou I. Molecules, 2009,14(8), 2729-34. https://doi.org/10.3390/molecules14082729.
CrossRef - Afifi, T. H., Okasha, R. M., Ahmed, H. E. A., Ilaš, J., Saleh, T., & Abd-El-aziz, A. S. EXCLI J., 2017, 16, 868–902. https://doi.org/10.17179/excli2017-356.
- Edraki, N., Iraji, A., Firuzi, O., Fattahi, Y., Mahdavi, M., Foroumadi, A., Khoshneviszadeh, M., Shafiee, A., & Miri, R. J. Iran. Chem. Soc., 2016, 13(12), 2163–2171. https://doi.org/10.1007/s13738-016-0934-7.
CrossRef - Parthiban, A., Kumaravel, M., Muthukumaran, J., Rukkumani, R., Krishna, R., & Rao, H. S. P. Med. Chem. Res., 2016, 25(7), 1308–1315. https://doi.org/10.1007/s00044-016-1569-z.
CrossRef - Angelova, V. T., Vassilev, N. G., Nikolova-Mladenova, B., Vitas, J., Malbaša, R., Momekov, G., Djukic, M., & Saso, L. Med. Chem. Res., 2016, 25(9), 2082–2092. https://doi.org/10.1007/s00044-016-1661-4.
CrossRef - El-Agrody, A. M., Abd El-Mawgoud, H. K., Fouda, A. M., & Khattab, E. S. A. E. H. Chem. Pap., 2016, 70(9), 1279–1292. https://doi.org/10.1515/chempap-2016-0049.
CrossRef - Refat, H. M., Fadda, A. A., & Kamal, S. J. Iran. Chem. Soc., 2015, 12(5), 845–854. https://doi.org/10.1007/s13738-014-0547-y.
CrossRef - Parthiban, A., Kumaravel, M., Muthukumaran, J., Rukkumani, R., Krishna, R., & Surya Prakash Rao, H. Med. Chem. Res., 2015, 24(3), 1226–1240. https://doi.org/10.1007/s00044-014-1190-y.
CrossRef - Hussain, M. K., Ansari, M. I., Yadav, N., Gupta, P. K., Gupta, A. K., Saxena, R., Fatima, I., Manohar, M., Kushwaha, P., Khedgikar, V., Gautam, J., Kant, R., Maulik, P. R., Trivedi, R., Dwivedi, A., Kumar, K. R., Saxena, A. K., & Hajela, K. RSC Adv., 2014, 4(17), 8828–8845. https://doi.org/10.1039/c3ra45749d.
CrossRef - Rahmani-Nezhad, S., Safavi, M., Pordeli, M., Ardestani, S. K., Khosravani, L., Pourshojaei, Y., Mahdavi, M., Emami, S., Foroumadi, A., & Shafiee, A. Euro. J. Med. Chem., 2014, 86, 562–569. https://doi.org/10.1016/j.ejmech.2014.09.017.
CrossRef - Choi, M., Hwang, Y. S., Kumar, A. S., Jo, H., Jeong, Y., Oh, Y., Lee, J., Yun, J., Kim, Y., Han, S. B., Jung, J. K., Cho, J., & Lee, H. Bioorg. and Med. Chem. Lett., 2014, 24(11), 2404–2407. https://doi.org/10.1016/j.bmcl.2014.04.053.
CrossRef - Ratnakar Reddy, K., Sambasiva Rao, P., Jitender Dev, G., Poornachandra, Y., Ganesh Kumar, C., Shanthan Rao, P., & Narsaiah, B. Bioorg. and Med. Chem. Lett., 2014, 24(7), 1661–1663. https://doi.org/10.1016/j.bmcl.2014.02.069.
CrossRef - El-Agrody, A. M., Fouda, A. M., & Al-Dies, A. A. M. Med. Chem. Res., 2014, 23(6), 3187–3199. https://doi.org/10.1007/s00044-013-0904-x.
CrossRef - El-Agrody, A. M., Fouda, A. M., & Khattab, E. S. A. E. H. Med. Chem. Res., 2013, 22(12), 6105–6120. https://doi.org/10.1007/s00044-013-0602-8.
CrossRef - Abd El-Wahab, A. H. F., Borik, R. M. A., Al-Dies, A. M., Fouda, A. M., Mohamed, H. M., El-Eisawy, R. A., Mora, A., El-Nassag, M. A. A., Abd Elhady, A. M., Elhenawy, A. A., El-Agrody, A. M. Sci. Rep., 2024, 14(1). 7589. https://doi.org/10.1038/s41598-024-56197-w.
CrossRef - Kandeel, M. M., Kamal, A. M., Abdelall, E. K. A., & Elshemy, H. A. H. Eur. J. Med. Chem., 2013, 59, 183–193. https://doi.org/10.1016/j.ejmech.2012.11.011.
CrossRef - Szulawska-Mroczek, A., Szumilak, M., Szczesio, M., Olczak, A., Nazarski, R. B., Lewgowd, W., Czyz, M., & Stanczak, A. Arch. Der Pharm., 2013, 346(1), 34–43. https://doi.org/10.1002/ardp.201200220.
CrossRef - Das, S. G., Doshi, J. M., Tian, D., Addo, S. N., Srinivasan, B., Hermanson, D. L., & Xing, C. J. Med. Chem., 2009, 52(19), 5937–5949. https://doi.org/10.1021/jm9005059.
CrossRef - Malik, M. S., Ather, H., Asif Ansari, S. M., Siddiqua, A., Jamal, Q. M. S., Alharbi, A. H., Al-Rooqi, M. M., Jassas, R. S., Hussein, E. M., Moussa, Z., Obaid, R. J., & Ahmed, S. A. Pharma., 2023, 16(3), 1–13. https://doi.org/10.3390/ph16030333.
CrossRef - Sabouri, S., & Abaszadeh, M. Polycy. Arom. Comp., 2021, 41(3), 467–477. https://doi.org/10.1080/10406638.2019.1597381.
CrossRef - Fallah-Tafti, A., Tiwari, R., Nasrolahi Shirazi, A., Akbarzadeh, T., Mandal, D., Shafiee, A., Parang, K., & Foroumadi, A. Med. Chem., 2011, 7(5), 466–472. https://doi.org/10.2174/157340611796799258.
CrossRef - Albalawi, F. F., El-Nassag, M. A. A., El-Eisawy, R. A., Mohamed, M. B. I., Fouda, A. M., Afifi, T. H., Elhenawy, A. A., Mora, A., El-Agrody, A. M., & El-Mawgoud, H. K. A. Int. J. Mol. Sci., 2023, 24(1), 49. https://doi.org/10.3390/ijms24010049.
CrossRef - Fouda, A. M., Irfan, A., Al-Sehemi, A. G., & El-Agrody, A. M. J. Mol. Str., 2021, 1240, 130542. https://doi.org/10.1016/j.molstruc.2021.130542.
CrossRef - Alblewi, F. F., Okasha, R. M., Hritani, Z. M., Mohamed, H. M., El-Nassag, M. A. A., Halawa, A. H., Mora, A., Fouda, A. M., Assiri, M. A., Al-Dies, A. A. M., Afifi, T. H., & El-Agrody, A. M. Bioorg. Chem., 2019, 87, 560–571. https://doi.org/10.1016/j.bioorg.2019.03.059.
CrossRef - Mohamed, H. M., Fouda, A. M., Khattab, E. S. A. E. H., El-Agrody, A. M., & Afifi, T. H. Zeit. Fur Natur. – Section C J. Biosci., 2017, 72(5–6), 161–171. https://doi.org/10.1515/znc-2016-0139.
CrossRef - Liu, X. H., Liu, H. F., Chen, J., Yang, Y., Song, B. A., Bai, L. S., Liu, J. X., Zhu, H. L., & Qi, X. B. Bioorg. and Med. Chem. Lett., 2010, 20(19), 5705–5708. https://doi.org/10.1016/j.bmcl.2010.08.017.
CrossRef - Wang, Y., Cheng, F. X., Yuan, X. L., Tang, W. J., Shi, J. B., Liao, C. Z., & Liu, X. H. Eur. J. Med. Chem., 2016, 112, 231–251. https://doi.org/10.1016/j.ejmech.2016.02.009.
CrossRef - Wu, X. Q., Huang, C., Jia, Y. M., Song, B. A., Li, J., & Liu, X. H. Eur. J. Med. Chem., 2014, 74, 717-725. https://doi.org/10.1016/j.ejmech.2013.06.014.
CrossRef - Di Braccio, M., Grossi, G., Roma, G., Marzano, C., Baccichetti, F., Simonato, M., & Bordin, F. Farm., 2003, 58(11), 1083–1097. https://doi.org/10.1016/S0014-827X(03)00160-5.
CrossRef - Hermanson, D., Addo, S. N., Bajer, A. A., Marchant, J. S., Das, S. G. K., Srinivasan, B., Al-Mousa, F., Michelangeli, F., Thomas, D. D., LeBien, T. W., & Xing, C. Mole. Pharma., 2009, 76(3), 667–678. https://doi.org/10.1124/mol.109.055830.
CrossRef - Tian, D., Das, S. G., Doshi, J. M., Peng, J., Lin, J., & Xing, C. Can. Lett., 2008, 259(2), 198–208. https://doi.org/10.1016/j.canlet.2007.10.012.
CrossRef - Das, S. G., Srinivasan, B., Hermanson, D. L., Bleeker, N. P., Doshi, J. M., Tang, R., Beck, W. T., & Xing, C. J. Med. Chem., 2011, 54(16), 5937–5948. https://doi.org/10.1021/jm200764t.
CrossRef - Mooring, S. R., Jin, H., Devi, N. S., Jabbar, A. A., Kaluz, S., Liu, Y., Van Meir, E. G., & Wang, B. J. Med. Chem., 2011, 54(24), 8471–8489. https://doi.org/10.1021/jm201018g.
CrossRef - Schmidt, J. M., Tremblay, G. B., Pagé, M., Mercure, J., Feher, M., Dunn-Dufault, R., Peter, M. G., & Redden, P. R. J. Med. Chem., 2003, 46(8), 1289–1292. https://doi.org/10.1021/jm034007d.
CrossRef - Chang, D. J., An, H., Kim, K. S., Kim, H. H., Jung, J., Lee, J. M., Kim, N. J., Han, Y. T., Yun, H., Lee, S., Lee, G., Lee, S., Lee, J. S., Cha, J. H., Park, J. H., Park, J. W., Lee, S. C., Kim, S. G., Kim, J. H., … Suh, Y. G. J. Med. Chem., 2012, 55(24), 10863–10884. https://doi.org/10.1021/jm301488q.
CrossRef - Wang, X., Nakagawa-Goto, K., Bastow, K. F., Don, M. J., Lin, Y. L., Wu, T. S., & Lee, K. H. J. Med. Chem., 2006, 49(18), 5631–5634. https://doi.org/10.1021/jm060184d.
CrossRef - Dong, Y., Shi, Q., Pai, H. C., Peng, C. Y., Pan, S. L., Teng, C. M., Nakagawa-Goto, K., Yu, D., Liu, Y. N., Wu, P. C., Bastow, K. F., Morris-Natschke, S. L., Brossi, A., Lang, J. Y., Hsu, J. L., Hung, M. C., Lee, E. Y. H. P., & Lee, K. H. J. Med. Chem., 2010, 53(5), 2299–2308. https://doi.org/10.1021/jm1000858.
CrossRef - Chandra, V., Fatima, I., Saxena, R., Kitchlu, S., Sharma, S., Hussain, M. K., Hajela, K., Bajpai, P., & Dwivedi, A. Amer. J. Obst. and Gyn., 2011, 205(4), 362.e1-362.e11. https://doi.org/10.1016/j.ajog.2011.05.024.
CrossRef - Griffin, R. J., Fontana, G., Golding, B. T., Guiard, S., Hardcastle, I. R., Leahy, J. J. J., Martin, N., Richardson, C., Rigoreau, L., Stockley, M., & Smith, G. C. M. J. Med. Chem., 2005, 48(2), 569–585. https://doi.org/10.1021/jm049526a.
CrossRef - Mun, J., Jabbar, A. A., Devi, N. S., Liu, Y., Van Meir, E. G., & Goodman, M. M. Bioorg. and Med. Chem., 2012, 20(14), 4590–4597. https://doi.org/10.1016/j.bmc.2012.04.064.
CrossRef - Burlison, J. A., Avila, C., Vielhauer, G., Lubbers, D. J., Holzbeierlein, J., & Blagg, B. S. J. J. Org. Chem., 2008, 73(6), 2130–2137. https://doi.org/10.1021/jo702191a.
CrossRef - Donnelly, A. C., Mays, J. R., Burlison, J. A., Nelson, J. T., Vielhauer, G., Holzbeierlein, J., & Blagg, B. S. J. J. Org. Chem., 2008, 73(22), 8901–8920. https://doi.org/10.1021/jo801312r
CrossRef - Le Bras, G., Radanyi, C., Peyrat, J. F., Brion, J. D., Alami, M., Marsaud, V., Stella, B., & Renoir, J. M. J. Med. Chem., 2007, 50(24), 6189–6200. https://doi.org/10.1021/jm0707774.
CrossRef - Yu, X. M., Shen, G., Neckers, L., Blake, H., Holzbeierlein, J., Cronk, B., & Blagg, B. S. J. J. Am. Chem. Soc., 2005, 127(37), 12778–12779. https://doi.org/10.1021/ja0535864.
CrossRef - Burlison, J. A., & Blagg, B. S. J. Org. Lett., 2006, 8(21), 4855–4858. https://doi.org/10.1021/ol061918j.
CrossRef - Nasr, T., Bondock, S., & Youns, M. Euro. J. Med. Chem., 2014, 76, 539–548. https://doi.org/10.1016/ j.ejmech.2014.02.026.
CrossRef - Bailly, C., Bal, C., Barbier, P., Combes, S., Finet, J. P., Hildebrand, M. P., Peyrot, V., & Wattez, N. J. Med. Chem., 2003, 46(25), 5437–5444. https://doi.org/10.1021/jm030903d.
CrossRef - Dong, Y., Nakagawa-Goto, K., Lai, C. Y., Morris-Natschke, S. L., Bastow, K. F., & Lee, K. H. Bioorg. and Med. Chem. Lett., 2010, 20(14), 4085–4087. https://doi.org/10.1016/j.bmcl.2010.05.079.
CrossRef - Dong, Y., Nakagawa-Goto, K., Lai, C. Y., Morris-Natschke, S. L., Bastow, K. F., & Lee, K. H. Bioorg. and Med. Chem. Lett., 2011, 21(1), 546–549. https://doi.org/10.1016/j.bmcl.2010.10.074.
CrossRef
Accepted on: 11 Mar 2025
Second Review by: Dr. Asif khan
Final Approval by: Dr. Abdelwahab Omri







ISSN Online: 2231-5039

![]()




{kind=link}