Molecular Docking-Based Approach to Elucidate the Efficacy and Toxicity of a Synthetic Biopolymer 1-(Carboxyamino) Ethyl Carbamic Acid for Use as a Coating Material on Cardiac Stents
 and Deepak Hujare
and Deepak HujareDepartment of Mechanical Engineering, Dr.Vishwanath Karad MIT World Peace University, Pune, Maharashtra India.
*Corresponding author E-mail: chopadejv91@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/420309
Download this article as:
![]()
The current study explores the potential of a novel synthetic biopolymer, 1-(carboxyamino) ethyl carbamic acid (CECA), which possesses hydrophilic and bioadhesive properties favorable for vascular environments. Molecular docking and computational analysis provide early-stage insights into potential bioactivity and safety of novel compounds. This work focuses on CECA’s interaction with cardiovascular-relevant targets and its predicted safety profile. This study aims to investigate the efficacy and potential toxicity of a synthetic biopolymer derived 1-(carboxyamino) ethyl carbamic acid as a coating material for cardiac stents using a molecular docking-based in silico approach. The synthesized polymer's molecular interactions with key cardiovascular proteins, Results suggest favorable binding affinities toward endothelial healing proteins and minimal interaction with RBC and toxicity targets, indicating promising biocompatibility. The findings support the potential of this polymer as a non-toxic, bio functional stent coating for improved cardiovascular implant performance.
KEYWORDS:Biopolymer; Cardiac stent; Coating material; Cytochrome-C Endothelial healing; 1-(carboxyamino) ethyl carbamic acid; Molecular docking; Toxicity
Introduction
Cardiac stent coating materials are critical for improving biocompatibility and minimizing thrombosis and restenosis1. This study investigates the efficacy and toxicity of the synthetic biopolymer 1-(carboxyamino) ethyl carbamic acid (CECA) as a potential cardiac stent coating using in silico approaches2. Molecular docking was performed against key cardiac-related protein targets. The long-term success of cardiac stents is often limited by restenosis and thrombosis, primarily due to poor endothelialization and inflammatory responses induced by metallic or poorly biocompatible coatings. Biopolymers have emerged as promising alternatives due to their tunable bioactivity and degradation profiles3-6. In this regard, 1-(carboxyamino)ethylcarbamic acid, a synthetic biopolymer, was suggested as a possible coating substance. In order to predict toxicity using pertinent in silico models and examine the biopolymer’s interaction with biological targets implicated in inflammation, thrombosis, and endothelial repair, this study employs computational molecular docking7.
Experimental
Material and methods
Ligand Preparation
The biopolymer structure based on 1-(carboxyamino) ethyl carbamic acid was built using ChemSketch optimized using MMFF94 force field8.
Protein Target Selection
Key protein targets were selected for evaluating efficacy and toxicity: Cytochrome C 1HRC
Molecular Docking and ADMET and Toxicity Prediction
AutoDock Vina was used for molecular docking, while PyRx was used for validation. The strength of the interaction was predicted using binding affinity ratings9-10.
Results and Disussion
Synthesis of Urethane derivatives
 |
Scheme 1 Click here to View Scheme |
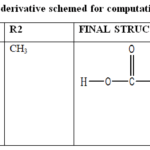 |
Table 1: List of derivative schemed for computational analysis Click here to View Table |
Docking with Endothelial Targets
The target protein selected for the study is Cytochrome C, with Protein ID 1HRC. The experimental structure was determined using X-ray diffraction, which aligns with the standard method commonly used for high-resolution structural analysis. No mutations were present in the protein, ensuring that the natural conformation was analyzed. PDB validation of the structure was rated as “better,” confirming its reliability and structural quality. Notably, there was no co-crystallized ligand in the structure12.In the Ramachandran plot. It sheds light on the protein backbone’s sterically permitted areas and structural preferences.
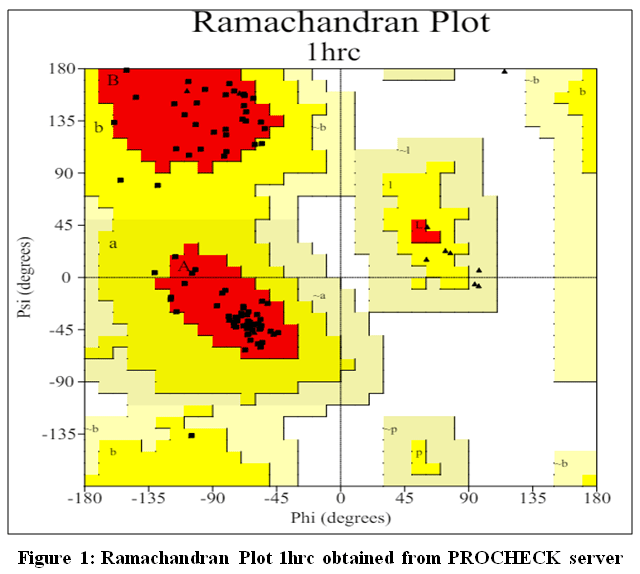 |
Figure 1: Ramachandran Plot 1hrc obtained from PROCHECK server Click here to View Figure |
Grid generation
Chimera 1.11.2, Maestro version 12.7.1619, and Auto Dock Tools 1.5.6 were used for grid generation and validation. When the co-crystallized ligand was available, its orientation was used to establish the grid parameters. The CASTp6 server helped with active site identification for proteins in the apo form, guaranteeing precise binding pocket selection. The grid that was created for the protein with PDB ID 1HRC included the residues of the active site, In order to guarantee the best possible representation of the binding pocket for next docking simulations, these residues were carefully chosen13.
Grid Parameter Optimization for Molecular Docking
To guarantee precise and effective ligand binding predictions in molecular docking experiments, the grid box must be defined correctly. For docking simulations, the enclosed grid box was meticulously adjusted to match the unique structural characteristics of the protein’s active site. Focused docking within the physiologically important region of the receptor was ensured by choosing the grid’s dimensions and placement to match the expected size and conformation of the ligands. Horseradish Peroxidase (PDB ID: 1HRC) was the protein employed in this investigation. The grid center was exactly located in the geometrical center of the protein’s active site, which is x = 44.969, y = 25.566, and z = 8.384. Based on past understanding of the catalytic pocket and the residues involved in ligand binding, this decision was made. In order to balance computational efficiency and biological correctness, the grid box dimensions were uniformly adjusted to 20 × 20 × 20 Å along the x, y, and z axes. This was done to account for the expected size of the ligands. This size minimizes the inclusion of irrelevant protein structural areas while guaranteeing that the grid completely encloses the active site14.
Ligand Preparation
ChemSpider, a reliable chemical structure database, provided the ligand structures. To guarantee ideal geometry and structural accuracy, these constructions were first cleaned in both 2D and 3D formats after being imported into MarvinSketch. The MMFF94 (Merck Molecular Force Field 94) was used to minimize the energy of the ligands during the cleaning process. Through the reduction of internal strain and steric conflicts, this minimization procedure aids in the achievement of thermodynamically stable conformations.The lowest energy conformer, which represents the most stable and physiologically significant form of the molecule, was chosen for further uses based on the energy minimization results. These completed ligand structures were exported in MOL2 format, which is appropriate for additional computational analysis and molecular docking15.
Interaction between certain ligands and target proteins
To assess the interaction between certain ligands and target proteins, molecular docking studies were carried out. First, the PDBQT format was created using the three-dimensional structures of the proteins and ligands. A unique in-house bash script that used ADFRsuite for receptor preparation and AutoDock Tools 1.5.6 for ligand preparation was used to accomplish this. While the receptor molecules were regarded as rigid during the docking simulations, all rotatable bonds within the ligands were maintained flexible, allowing them to explore multiple conformations. AutoDockVina v1.2.3 was used to perform docking calculations using a grid spacing of 0.375 Å. The search was expanded to look for possible other binding sites for improved interaction mapping, with the grid box centered on the target protein’s known active site. Unless otherwise noted, all other docking parameters were kept at their default settings. The docking protocol was configured as follows: CPU threads: 23, Exhaustiveness: 32,Number of binding modes: 9,Energy range: 3 kcal/mol. The XYZ coordinates of the grid center that is utilized for docking. Re-docking studies were carried out in the exact same settings as the original docking runs in order to guarantee the docking protocol’s dependability and reproducibility.
Visualization
Using Biovia Discovery Studio Visualizer, post-docking results from AutoDockVinawere further examined to produce ligand–protein complexes. Key binding interactions were visually interpreted for these complexes. To show the binding conformation and interaction profile of ligands within the target protein’s active site, Maestro 12.3 (Schrödinger, Academic Edition) was used to generate 2D interaction diagrams and 3D structural representations of the docked complexes. The illustrations included thorough explanations of hydrophobic contacts, hydrogen bonds, and other important chemical interactions.
Table 2: Binding Energy and Interaction
|
Complex |
Binding Energy | Type of Interaction | Residue ID |
Distance |
|
1_1HRC |
-5.007 | Hydrogen Bonds | GLY23A | 2.06 |
| ASN31A | 3.37 | |||
| ASN31A |
2.54 |
|||
| ARG38A |
2.33 |
|||
| ARG38A |
2.21 |
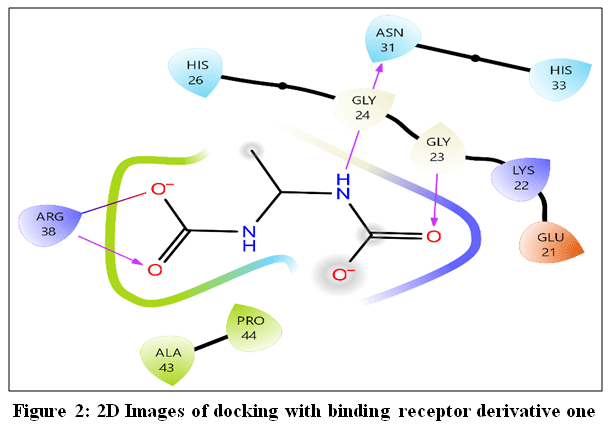 |
Figure 2: 2D Images of docking with binding receptor derivative one Click here to View Figure |
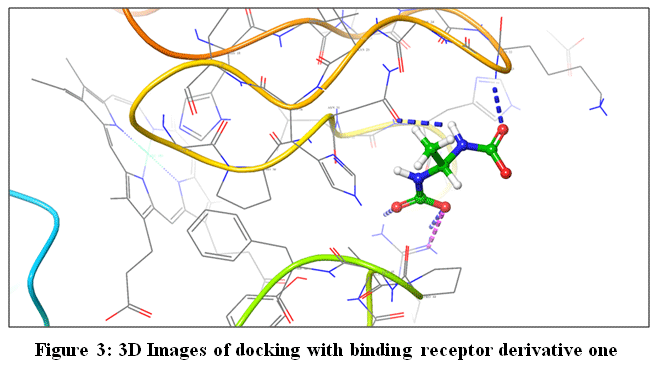 |
Figure 3: 3D Images of docking with binding receptor derivative one Click here to View Figure |
Toxicity Profiling
Methodical Process
Step I: Getting inside the Software Go to https://www.swissadme.ch in a web browser.
Step 2. Installation and registration are not necessary.
Step 3: Compound Input was given as export the chemical structure in smiles format after drawing it with ChemSketch, into the SwissADME website’s input box.
Step 4: Click “Run” once the SMILES strings have been entered. Usually, the software takes a few seconds to several minutes to process the input16 and produce results shown in figure 4.
 |
Figure 4: Pharmacokinetics study of 1-(carboxyamino) ethyl carbamic acid Click here to View Figure |
| Parameter | Level |
| GI absorption | High |
| BBB permeant | No |
| P-gp substrate | No |
| CYP1A2 inhibitor | No |
| CYP2C19 inhibitor | No |
| CYP2C9 inhibitor | No |
| CYP2D6 inhibitor | No |
| CYP3A4 inhibitor | No |
| Log Kp (skin permeation) | -6.67 cm/s |
Low cardiotoxicity risk is indicated by little binding to site and no significant binding to CYP450 enzymes (>−5.0 kcal/mol). ProTox-II indicates a low likelihood of mutagenicity or hepatotoxicity, therefore favorable prospects for drug development are compounds that exhibit good GI absorption, no CYP inhibition, adherence to Lipinski’s guidelines, and no pains alarms.
Conclusion
The biopolymer’s 1-(carboxyamino) ethylcarbamic acid derivatives as potential, efficient and bioinert stent coating material is supported by the in silico analysis. Its reduced toxicity profile and favorable interactions with endothelial targets enhance its suitability for cardiovascular applications. It having good binding energy on selected protein target site. These derivative can be use as a coating for cardiac stents. The synthetic biopolymer made from 1-(carboxyamino) ethylcarbamic acid exhibits a safe toxicity profile and encouraging biological interactions. Nevertheless, urethane derivatives for covering cardiac stents have been successfully developed and validated experimentally using cytotoxicity tests as a vitro models.
Acknowledgment
The authors acknowledge support from Vineet analytical Research Laboratories Pvt Ltd. Pune for technical assistance in docking research.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Ba-Ghazal H, Yousef T. A, Bedier R. A, Al-Janabi S. A. M, Alaasar M, Al-Duaij O. K and Shaaban S. Synthesis, Characterization, DFT and Molecular Docking Analysis of N-phenyl-2-((4-(3-phenylthioureido)phenyl)selanyl) acetamide. Orient J Chem 2025, 41(2), 654-664
CrossRef - Angel Praba, P. Christina Ruby Stella and A. Nive. In silico and In vitro Analysis of Justiciaadhatoda Methanol Extract: Molecular docking Insights and A Promising Approach for Drug Discovery. Orient J Chem. 2025, 41(2), 498-505
CrossRef - Kelly Navas-Gomez and Manuel F. Valero, Why Polyurethanes Have Been Used in the Manufacture and Design of Cardiovascular Devices: A Systematic Review, 2020,1,167-172
CrossRef - Prabhakar G, Madhukar G. V. R. S, Domala R. Synthesis, Characterization, Biological Assay of New 5-(Pyridine-2-Yl)-1,3,4-Oxadiazol-2-Amine Derivatives and their Molecular Docking Studies. Orient J Chem 2024;40(3),737-743
CrossRef - David Wienen A, Thomas Gries A, Stuart L. Cooper b, Daniel E. Heath, An overview of polyurethane biomaterials and their use in drug delivery, Journal of Controlled Release, 2023, 3,376-388,.
CrossRef - RanaM, Ghosh N. S, Kumar D, Singh R andMonga J. 2D-QSAR Assisted Design, and Molecular Docking of Novel IndoleDerivates as Anti-Cancer Agents. Orient J Chem 2024;40(5), 1440-1448
CrossRef - Gawade V andChopadeV. V. Design and Synthesis of Novel Series of Thiophene-2,5-dicarbohydrazide Derivatives as Potential Anticancer Agents. Indian J of Pharmaceutical Education and Research.2024;58(3):837-53.
CrossRef - Sutradhar R. K, Marma A and Hossain M. E. Synthesis, Bioactivity Screening and Docking Analysis of Thiazole Derivatives Containing Quinoline Moieties. Orient J Chem 2024;40(5),1297-1305
CrossRef - Chopade J.V. and Hujare D. P. Biocompatibility and Haemocompatibility Assessments of RBC with Polymeric Coated Cardiac Stent. International Journal of Pharmaceutical Quality Assurance.2024;15(3):1650-1653.
CrossRef - Ali, A., Mir, G. .and Ayaz, A.Insilico analysis and molecular docking studies of natural compounds of Withaniasomnifera against bovine NLRP9. J Mol Model, 2023, 29, 171-176.
CrossRef - Banpure S.G., Chopade V.V. and Chaudhari P.D. Anti-Alzhimer Activity of Bay Leaves in Scopolamineinduced Rat Model.International Journal of Drug Delivery Technology.2023;13(1):17-22.
CrossRef - Mishra S, Sachdeva M, Nimesh H. Network Pharmacology and Molecular Docking to Identify the Molecular Targets of Novel Indole-Quinoline Derivative in Cancer. Orient J Chem 2025;41(1), 217-229.
CrossRef - Chopade J.V, Hujare D. P. Characterization and In-vitro Study of Polyethylene Glycol as Coating Material used as Drug Carriers on Coronary Stent for Treatment of Cardiac Diseases. International Journal of Drug Delivery Technology.2024;14(2):955-960.
CrossRef - Sonia Fathi-Karkan, BehazBanimohamad-Shotobani, SepidehSaghati, Reza Rahbarghazi and SoodabehDavaran “A critical review of fibrous polyurethane-based vascular tissue engineering scaffolds”, Journal of Biological Engineering, 2022, (2),263-271.
CrossRef - Agu, P.C., Afiukwa, C.A., Orji, O.U. et al. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci Rep, 2023, 3,483-487
CrossRef - Rathi M. V. Revealing the Toxicity of Chlorate through the Analysis of Molecular Interaction Using Viscosity Measurements and Apparent Molar Volumes. Orient J Chem 2021;37(6),1336-1343
CrossRef
Accepted on: 27 Feb 2026
Second Review by: Dr. Jennicav Jove
Final Approval by: Dr. B.K Sharma







ISSN Online: 2231-5039

![]()




{kind=link}