Advances in Analytical Techniques for Anticancer Drugs: Method Development, Validation and Insights into Newly Approved Therapies
 , Nallala Ahalya, Maskuri Niharika, Manne Sai Jayachandra and Jahnavi Bandla*
, Nallala Ahalya, Maskuri Niharika, Manne Sai Jayachandra and Jahnavi Bandla*Department of Pharmaceutical Analysis, Vishnu Institute of Pharmaceutical Education and Research, Narsapur – 502313, Medak, Telangana, India.
Corresponding Author Email: jahnavi.bandla@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/420216
Download this article as:
![]()
The continuous evolution of analytical techniques has significantly enhanced the development, validation, and regulatory compliance of methods for anticancer drugs. This review highlights the latest advancements in both chromatographic, spectroscopic, as well as hyphenated methods applied to the examination of anticancer agents. Special emphasis is placed on newly approved therapies, including Revumenib, Capivasertib, Pirtobrutinib, Pacritinib, and Pralsetinib, which have demonstrated promising clinical outcomes. Method development and validation strategies for these drugs are discussed in the context of pharmaceutical quality control and bioanalytical applications. Additionally, the review explores emerging trends in analytical methodologies, such as high-resolution mass spectrometry and novel sample preparation techniques, which contribute to improved sensitivity, specificity, and efficiency. These advances facilitate precise quantification, stability assessment, and pharmacokinetic profiling of anticancer agents, ultimately aiding in optimized therapeutic outcomes.
KEYWORDS:Anticancer Drugs; Analytical Techniques; Capivasertib; Method Development; Pirtobrutinib; Pacritinib; Pralsetinib; Revumenib
Introduction
Cardiovascular disorders are the world’s largest cause of death, followed by cancer and has claimed millions of lives. According to a 2014 report from the World Health Organization (WHO), worldwide cancer 2008 saw 12.7 million cases, whereas 2012 saw 14.1 million cases. In 2018, deaths from cancer reached 9.6 million, with 18.1 million new cases reported. One characteristic of cancer is the uncontrollable proliferation of cells, leading to the formation of either solid or liquid tumors1.
The primary treatment approaches for cancer include chemotherapy, radiation therapy, and surgery. Chemotherapy aims to eliminate or significantly suppress tumor cell growth and involves various classes of anticancer drugs, like microtubule inhibitors, hormones, hormone antagonists, alkylating compounds, antibiotics, and antimetabolites2. Conventional anticancer drugs exhibit high reactivity, leading to significant cytotoxic effects and adverse reactions. Their inherent toxicity and instability necessitate stringent quality and safety measures. Consequently, analytical techniques have a vital part in the anticancer medication monitoring of therapeutic medications3.
The analysis of anticancer drugs in biological samples serves three primary purposes: therapeutic drug monitoring, drug development, and exposure assessment for healthcare professionals handling these agents. Various human biological matrices, including cerebrospinal fluid(CSF), tissue, blood, urine, and saliva, are commonly used for drug monitoring.
Analytical chemistry plays a fundamental role in drug quality control, confirming safety and effectiveness of anticancer medications. To quantify anticancer medications in biological samples, a numerous analytical methods were studied in this literature. These techniques include capillary electrophoresis (CE), gas chromatography (GC), gaschromatographymassspectrometry/mass spectrometry (GC-MS), reversephasehighperformanceliquidchromatography (RPHPLC), ultraviolet spectrophotometry (UV), Fouriertransforminfraredspectroscopy (FTIR), gaschromatographymassspectrometry (GCMS), GC MS-MS, and GC MS/MS drug analysis also monitoring, ultimately contributing to improved cancer treatment outcomes.
Revumenib
Revumenib (RVB) is used to treat acute leukemias that are KMT2A-rearranged (KMT2Ar), also referred to as SNDX-5613, binds to the receptive site of menin and displaces KMT2A. The HOX and MEIS genes are successfully shut down by this action, which stops the growth of leukemic cells. Revumenib was developed by SyndaxPharmaceuticalsInc(Waltham MA USA) (Fig.1). Acute leukemias that have revert or recalcitrant (R/R) KMT2A rearrangements, such as NPM1-mutant AML,acutemyeloidleukemia(AML) as well as acutelymphoblastic leukemia(ALL), are being administered using RVB, a pioneering menin inhibitor. For the medical healing of patients with AML, RVB has been designated as an Orphan Drug by the US FDA. Additionally, the FDA has given it Fast Track status for treating R/R acute leukemias in adults and children with a KMT2A realignment or NPM1 mutation. The FDA has awarded RVB Break through Therapy Designation for curing R/R acute leukemia among children as well as in adults with a KMT2Ar4. The FDA has given RVB’s new drug application (NDA) priority review status. The FDA’s RealTimeOncologyReview Program (RTOR) is now assessing the NDA submission in accordance with Prescription Drug User Fee Act (PDUFA), and September 26, 2024 has been set as the target action date5.
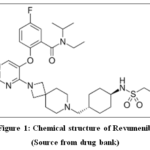 |
Figure 1: Chemical structure of Revumenib Click here to View Figure |
(Source from drug bank)
Using an ESI source, Mohamed W. Attwa and colleagues (2024) sought to develop a quick, accurate, environmentally friendly, and RVB proportion in human liver microsomes (HLMs) can be estimated using the hypersensitive UPLC-MS/MS methodology. The validation methods were conducted in compliance with the USFDA’s validation standards towards biological analysis techniques, which include matrix effect, extraction recovery, linearity, selectivity, precision, accuracy, and stability. The authentication properties of UPLC MS/MS technique yielded findings that met FDA standards. RVB precursor ion were generated by the positive ESI origin, also their two daughter ions were evaluated by means of MultipleReactionMonitoring(MRM) means. Revumenib and encorafenib were separated by C8column(2.1mm,50mm, and3.5µm) with a constant eluent. From 1 to 3000 ng/mL, the RVB calibration curve’s linearity (y = 0.6515x − 0.5459andR2 = 0.9945) fluctuated. Closeness as well as the exactness, which varied as of -0.88% to 11.67% within a working day and values from -0.23% to 11.33% between days, validated the replicability of UPLC MS-MS analytical method. Vulnerability of the new technique verified that RVB levels could be measured at a LOQ of 0.96 ng/mL. Also current method’s greenness was confirmed by the AGREE score of 0.77. RVB is comparable to medications with a high extraction ratio, as evidenced by its quick invitro t1/2(14.93min)and elevated intrinsicclearance(54.31 mL/min/kg). The current LC-MS/MS method is thought to be the first analytical methodology that uses metabolic reliability measurement for Revumenib evaluation in HLMs. The techniques being crucial for furthering creation of novel medications, especially when it comes to improving metabolic stability6.
Capivasertib
The discovery of the pan-AKT kinase inhibitor capivasertib raises the possibility that treating breast cancer may eventually involve targeting the PIK3/AKT pathway7. In November 2023, capivasertib was given acceptance for its usage in US medicine8. The capivasertib’s fast track designation application was accepted by the FDA9. Capivasertib’sempirical formula is C21H25ClN6O2, and its molecular weight is 428.915, as illustrated in Fig. 210,11. According to the literature analysis, only two analytical procedures were developed: the HPLC method and liquidchromatographytandemmassspectrometry (LC-MS MS).
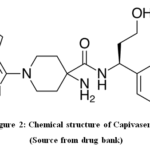 |
Figure 2: Chemical structure of Capivasertib Click here to View Figure |
(Source from drug bank)
In 2024, Bavita Gaur and associates used a recently developed and proven steadiness representing reversephasehighperformanceliquidchromatography {RP-HPLC} technology to measure Capivasertib utilize Waters Symmetry column having UV detection, a wavelength of 260 nm. It only takes five minutes to finish the analysis. Capivasertib was recovered and separated after 3.3475 minutes of retention. Capivasertib was shown to have a linear concentration range of 50–300 µg/mL.Capivasertib’s regression equations were found to be y = 13485.85x + 1723.04. Capivasertib was discovered that the quantification limits of capivasertib were 2.00 and 0.5000 μg/mL, however the limit of detection been 0.6 μg/mL12.
Takeo Yasu along with YoshitoGando (2024) suggested utilizing HPLC-UV to ascertain the concentration of capivasertib in human plasma. Methanol-induced protein precipitation was followed elution that is isocratic by on a C-18 column to distinct capivasertib and pirfenidone (internal standard). Pumped with constant rate of flow 1.0 ml/min, the phase of mobility consists of 0.5% KH2PO4 (pH 4.5)/acetonitrile in 73:27 (vol/vol) ratio. The measurement of quantity was done at 219 nm. The curves of calibration showed a straight-line trend between 50 and 1000 ng/ml. The coefficients of variation within and between days were less than 10.2%. The recovery was greater than 93.8%, and the assay accuracy was between 7.2-2.9%13.
Wen-Wu Cheng and colleagues (2020) developed a uncomplicated also exact liquid chromatography tandem mass spectrometric technique meant for measuring capivasertib in plasma of dog with ipatasertib as an internal reference. Acetonitrile was used to deproteinate the plasma samples. A 40°C-stored Acquity Bridged Ethyl Hybrid C18column (1.7μm,2.×50mm) applied in support to chromatographic separation. Acetonitrile, water, and 0.1%formicacid made up solvent. The MultipleReactionMonitoring transition for ipatasertibhas been m/z 458.2 >387.2, whereas with capivasertib, it was m/z 429.2 >135.1. The concentration range of the results showed excellent linearity of 1–1,000 ng/ml, having a correlation of coefficient of >0.9981. The quantitation’s inferior boundary be one ng/ml. Capivasertib was extracted from dog plasma with a recovery rate of >85.81%, and also no discernible the matrix effect was present. The precision within and between days was lower than 9.58%, while the accuracy range was from -10.60% to 12.50%. Using approved technique, the biodisposition analysis of capivasertib in plasma of dog following IV route (1 mg/kg) and single orally (5mg/kg) dose was carried out. As per the findings, capivasertib was quickly taken up by plasma, exhibiting low clearance and a high bioavailability of 47.04%14.
Pirtobrutinib
Mantle cell lymphoma is treated with the anticancer medication pirtobrutinib, which is sold under the Jaypirca brand. It limits growth and endurance of B cell lymphocytes by blocking Bruton’styrosinekinase (BTK). In November 2023, European Union also the US FDA approved use of one pirtobrutinib in medicine15,16. The most recurring adverse effects comprise musculoskeletal pain, tiredness, oedema, loose stool, and dyspnea17. When taking this medication to treat mild or chronic lymphocytic leukemia, the most frequent adverse effects are bruises, edema, nausea, pyrexia, headaches, extreme tiredness, tussis, soft tissue pain, diarrhea, pneumonia, stomach discomfort, and dyspnea18-21. The production of antibodies is the responsibility of one subtype of lymphocytes that is B cells which are the white blood cells.
Abandoned B cell progression can lead to cancer. The BTK enzyme is necessary for B cells to survive and proliferate. Pirtobrutinib inhibits BTK in a different way than the conventional BTK inhibitor ibrutinib. It accomplishes this by preventing a genetic transformation (change at Cysteineresidue C481 of active site of BTK) that could reduce the sensitivity of certain malignancies to ibrutinib. The USFDA approved pirtobrutinib in December 2023 for a list of disorders that now includes people with long-term lymphocytic leukemia22,23. The US FDA granted authorized utilization of Eli Lilly and company’s pirtobrutinib during January2023 for management of mantlecelllymphomahad developed a resistance to traditional BTK inhibitors.
Pirtobrutinib, a small chemical that prevents BTK in a very particular non-covalent way. As it was having magnificent selectivity, atrial fibrillation occurrence was linked to lower chance for quitting from adverseeffects. The amino acid that is cysteine 481 (Cys 481) in the active region of BTK is interacting with imatinib and also rest BTK covalent inhibitors; however, pirtobrutinib’s restraining act is not influenced by mutations in Cys 481. This resistance to covalent BTK inhibitors appears to be most commonly caused by Cys 481 mutations, while the correct mechanism behind this confrontation are yet unclear. Pirtobrutinib’s chemical formula is 5 amino 3[4[[(5fluoromethoxybenzoyl)amino], as [methyl]phenyl is shown in Fig. 3. In ((2S)1,1trifluoropropan2yl)1 [4carboxamidpyrazole]24
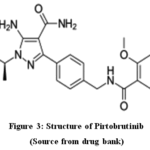 |
Figure 3: Structure of Pirtobrutinib Click here to View Figure |
(Source from drug bank)
LC-MS/MS was used by Hemavathi N. Deepakumari et al. (2024) to create, verify, and describe products of forced degradation. Pirtobrutinib was quantitatively measured using an isocratic HPLC approach at a λmax of 219 nm. The process that was employed was simple, clear, tested, and selective. For isocratic elution, the samples were passed through an Agilent Eclipse C18column (15×4.6 mm,3.5m). Flow rate being 1.0micro-Litre per minute, eluent which included 0.1%formicacid also acetonitrile was administered as 30:70 v/vratio. The range of concentration 0.0 to 150 mg mL-1, as response is linear. Pirtobrutinib’s quantitation and detection limits were determined to be 0.1 and 0.3, respectively. According to conventional ICH principles, the method’s precision, accuracy, linearity, robustness, and system applicability were evaluated. The results were determined to be within reasonable bounds. To test the method’s effectiveness and stability, the medicine was subjected to a range of stress conditions, including hydrolysis, oxidation, reduction, acids, alkalis, and photo- and thermal degradations. Significant degradation was seen in peroxide, reduction, acidic, and alkaline environments. During the forced degradation investigations, the products were obtained as degradation products and were analyzed and characterized using mass spectrometry. Therefore, pirtobrutinib can also be quantified using the suggested method when its degradation products are present. Pirtobrutinib is consistently quantified using this method due to its increased sensitivity and regulatory compliance25.
Dong R. et al. (2025) developed a reliable and strong technique for measuring pirtobrutinib inplasma of rat throughultrahighperformanceliquidchromatographytandemmassspectrometry(UHPLCMSMS).Zanubrutinib acted also as internal standard (IS),andAcetonitrile and 0.1%formicacid made up eluent. Ion quantification transitions for pirtobrutinib and zanubrutinib were m/z 480.12→294.05 and 472.20→ 289.96, respectively. Pirtobrutinib had relative standard deviations (RSD%) of less than 9.8% within a day and 10.3% between days. The ranges for matrix effects and recovery were 91.7–100.4% and 95.1–101.5%, respectively26.
Pacritinib
The chemical symbol for pacritinib is (16 E) [2-(1-Pyrrolidinyl) ethoxy]-11-[19.3.1.12,6.18,12]-14,19-dioxa-5,7,26-triazatetracycloheptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene. The empirical formula is C28H32N4O3 (Fig. 4). Myelofibrosis is a very rare disease that results in fibrosis and abnormalities in the hematopoietic cells of the bone marrow27. Although the exact origin of primary myelofibrosis (MF) is unknown, people with a history of polycythemiavera or essential thrombocythemias are more likely to develop secondary MF. MF can arise after essential thrombocythemias or polycythemiavera since it can be either primary or secondary. Mutations in JAK 2 have been linked to both types of MF, in spite of the statement that precise reason of MF is uncertain28,29.
The pharmacologic action of pacritinib is believed to be achieving via constraining wildtype JAK2, mutanJAK2V617F, also FMSlikeTyrosineKinase3 (FLT3). While this has no effect on JAK 1, it exhibits more inhibitory activity against JAK2 distinct from proteins that are comparable (such TYK 2 and JAK 3). At concentrations that are therapeutically relevant, it inhibits JAK2. Although it’s uncertain if this effect is therapeutically significant, pacritinib has some inhibitory activity against other cellular kinases, like IRAK 1 as well as CSF1R.
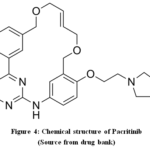 |
Figure 4: Chemical structure of Pacritinib Click here to View Figure |
(Source from drug bank)
In order to perform kinetic studies in hale and hearty rabbits, K. Ramakrishna et al. (2024) set out to create and validate a precise also selective LC ESI MS-MS methodology in favour of its measurement of pacritinib. Chromatographic resolution has been achieved using hypersil/ODS(50mm×4.6mm,3μ)Analytical C18Column with an eluent that included 0.1% formicacid and Acetonitrile 25:75 ratio.Solvent phase system was flowed from its analytical column with flow rate 0.6 milliliter per minute. Using this methodology, known ionic transitions of m/z-473.25/98.09 for pacritinib and 506.18/57.12 for the internal standard (Amprenavir) were tracked in multiplereactionmonitoring(MRM). The calibrationplot’sregressionline was y = 0.0002x+0.007, with a correction value (r2) of 0.9989. The CV results for the matrixeffectwere 4.79% on lowQC and 4.91% on highQC levels. In HighQC(12.70μg/ml), MQC(8.50μg/ml),and LowQC(1.19μg/ml), the average percentage recoveries for pacritinib were 95.87%, 103.64%, and 94.32%, respectively. QC(1.19,8.50,and12.70μg/ml) sample had findings ranging from 2.98 to 5.07%. Using the established procedure, the kinetics of pacritinib following oral ROA among rabbits were examined. Later giving pacritinib orally to rabbits that are healthy, pharmacokinetic factors was shown, also the recognized procedure was efficiently validated30.
PagalaBangaraiah and MulamrajuAruna Kumar (2024) set out to validate a precise and linear liquidchromatographyelectrosprayionizationtandemmassspectrometrytechnique in order to produce and quantify pacritinib. Using the Hypersil Gold C18 column, a chromatographic resolution of 50 × 4.6 mm, 2.1 µm was attained. The eluent consists about 20:15:65 (%v/v/v) methanol, 0.1%formicacid, and acetonitrile. This process involved closely observing the completed ionictransitions of m/z 473.1/376.2 in favour of pacritinib and 584.26/101.1 for internal standard for brigatinib in the multiple reaction monitoring mode. The linear plotregressionline, represented as y = 0.0001x − 0.0008, has a significant correlation value (r2) of 0.9997. When the quality control (QC) level was poor, the matrix effect’s relative standard deviation (RSD) was 3.63%; when it was high, it was 3.57%. Pacritinib’s percentage average recoveries during the study were 103.27% in high QC, 97.59% in medium QC, and 94.28% in low QC, in that order. The QC samples (0.378, 1.058, 7.56, and 11.34 µg/mL) yielded results ranging from 2.00% to 4.03%. In order to assess Pacritinib in biological samples for QC, forensics, and individual bioavailability studies, the devised approach proved beneficial31.
YaMeng Wu et al. (2023) created a quantitative LC-MSMS detection technique for pacritinib in plasma of rat utilizing Ibrutinib as an internal reference. Rat pharmacokinetic and drug-drug interaction studies were then conducted using this methodology. Within the extent of 1–1500 ng/mL, the established methodology shown great linearity by a lowerlimitofquantificationof1ng/mL. Additionally, Pacritinib’sintraandinter-day precision RSD% was less than 10.69%, and its range of accuracy was -2.31% to 2.08%. The stability, matrix effect, also recovery all complied by FDA regulations. This technique worked well for quantitatively determining the amount of pacritinib in rat plasma. When compared to voriconazole, we found that isavuconazole greatly slowed the metabolism of pacritinib in the pharmacokinetic experiments. As a result, AUC (0-t) increased by 2.5 times, AUC (0-∞) enlarged by 2.3 times, CLz/F improved by 4.4 times, and Cmax increased by 3.4 times. A trustworthy LC-MSMS technique for determining the concentration of drug in plasma of pacritinib among rats was successfully developed in this work. According to pharmacokinetic research, isavuconazole is more to be expected than voriconazole to raise the exposure of blood to pacritinib32.
Pralsetinib
Pralsetinib, a selective RET enzyme inhibitor, is used to treat adult individuals havinglocallyadvancedormetastaticnon-smallcelllungcancer(NSCLC) who have RET genefusionmutationssucceeding platinum-based chemotherapy. Pralsetinib’s chemical formula is ((R)-3-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl) (Fig. 5). -1-((1s,4S) 5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl) cyclohexyl-1-methoxy-4-(4-methyl-6-) ethan-1-amine butan-1-one–(S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl).In March 2021, the China NationalMedicalProductsAdministration approved, a highly selective RET inhibitor33, for treatingmetastatic or locallyadvancednon-smallcelllungcancer in adult or grown-up individuals having RETgene fusionwho already underwent platinum-based chemotherapy.
 |
Figure 5: Chemical structure of Pralsetinib Click here to View Figure |
(Source from drug bank)
Zhu Yonghong et al. (2024) constructed and validated the liquid chromatography methodology regarding the accurate identification of pralsetinib and related contaminants. Using a WatersXBridge C18column measuring 4.6mm by 250mm also having a particlesize around 5μm, pralsetinib and its related contaminants were separated. Using a gradient elution technique, eluent A having composition around 20mmol/L potassiumdihydrogenphosphate(KH2PO4) along with acetonitrile (ACN) having capacity ratio 19:1, whereas eluent B included solely ACN. For the detection, 260 nm as the wavelength, 10 μL as the injectionvolume, and 1.0 mL/min as its rate of flow were employed. This chromatographic methodology proposed here was verified in in confirmity with ICH Q2 “R1” requirements. This approach has showed great linearity in a particular range of concentration (imp-A: 0.035-10.21 μg/mL; imp-B: 0.09-10.16 μg/mL; imp-C: 0.15-10.19 μg/mL; pralsetinib: 0.04-10.32 μg/mL). The approach also shows strong sensitivity, with quantification limits of 0.035, 0.09, 0.05, and 0.04 μg/mL and detection limit of 0.01, 0.03, 0.015, and 0.013 μg/mL for pollutants A, B, C, and pralsetinib, respectively. The method’s selectivity, steadiness, reproducibility, exactness, and heftiness met the validation receiving standards34.
Yan Zhang et al. (2024) set out to introduce a methodology for detecting pralsetinib concentration in human’s plasma as well as cerebrospinal fluid (CSF) using UPLC-MSMS. This methodology having its correctness, exactness, steadiness, withdrawal recovery, and matrix effect all validated using the external standard method. Every working solution was made from stock solutions of pralsetinib that contained 1 mg/mL. The plasma or CSF samples have been broken up on an ACQUITY UPLC HSS T3column(2.1×100 mm, 1.8 μm) having gradientelution method after precipitated by acetonitrile for protein precipitation using 0.1% formic acid (solution A) and acetonitrile (solution B) as solvents at a rate of flow of 0.4 mL/min.All outcomes of the validation, which included stability, extraction recovery, matrix effect, precision, accuracy, selectivity, and calibration assessment, were deemed satisfactory. Pralsetinib levels in a clinical sample have been effectively identified using this technique; the concentrations in the plasma and CSF was discovered to be 61.55 μg/mL and 475 ng/mL. For clinical medication monitoring, we have emerged with a rapid and effectual technique for determining concentrations in the two, plasma as well as CSF. The method may also be used as a roadmap for further optimization35.
The goal of RushitaSardhara et al. (2024) about to set up an RP-HPLC methodology meant for pralsetinib measurement at a detection wavelength of 270 nm and at rate of flow 1 ml/min using BDSHypersilC18(250 mm × 4.6 mm,5µparticle size) with OPA buffer pH3.0: methanol (45:55). 80–100 µg/ml is the linearity range, and 99.81 ± 0.579–99.968 ± 0.291% is the accuracy percentage recovery Pralsetinib’s quantification limit (LOQ) is 7.07 µg/ml, while its limit of detection (LOD) is 2.34 µg/ml. No significant interferences in the determination are confirmed by the linearity, repeatability, and recovery results. Pralsetinib was reported to undergo considerable degradation in oxidative, photolytic, and acidic and alkaline conditions. Thus, this technique can be applied to regular quality control evaluations of this medication36.
A reliable HPLC approach for impurity analysis was presented by Rajesh VarmaBhupatiraju et al. in 2024. They also used LC-MS to describe degradation products (DP) and assess the method’s environmental impact. The study first improved HPLC settings using a range of columns and buffers in order to successfully separate by means of an X Bridge® RP-C18 column by using ethanol as mobile phase A and 50 mM formic acid at pH 2.9. Great peak resolution along with the symmetry were given by this configuration, which is necessary for trustworthy stability investigations. The sensitivity and detection efficiency of DPs were then improved by adapting the existing HPLC method for HPLC-MS/MS. Pralsetinib underwent considerable deterioration in acidic (29.3%) and basic (21.5%) environments, along with many DPs detected, according to studies of its stress degradation in different circumstances (acidic, basic, oxidative, thermal, and photolytic). Degradation process under thermal also with photolytic circumstances was minimal, while it was 19.8% under oxidative stress. HPLC-MS/MS study provided detailed information regarding the degradation and stability mechanisms of pralsetinib by determining the structures of five degradation products. The robustness, accuracy, precision, linearity, sensitivity, specificity, and selectivity of respective methodology were confirmed through method validation in accordance with ICH recommendations Q2(R1). With a coefficient of determination (r2) for pralsetinib and its impurities more than 0.999, the approach demonstrated strong linearity. This technique improves DPs characterization and impurity identification, guaranteeing pralsetinib’s quality and safety. In keeping with sustainable analytical standards, the method’s environmental impact was also evaluated. These results offer crucial information about the stability of pralsetinib, directing circumstances of storaging and guaranteeing its effectiveness as well as safety in medicinal applications37.
Conclusion
To make sure that the effectiveness, safety and regulatory compliance of anticancer medications, the development of analytical tools remains crucial. Drug characterization and bioanalysis have been greatly enhanced by usage of state-of-the-art techniques like tandemmassspectrometry(MS/MS), ultra-high-performanceliquidchromatography(UHPLC), and chemometric methods. The review emphasizes how crucial method validation parameters are to guaranteeing accuracy and reproducibility for recently approved anticancer drugs as pralsetinib, revumenib, capivasertib, pirtobrutinib, and pacritinib. Integration with automation and artificial intelligence is anticipated to substantially improve technique robustness and efficiency as analytical science advances. Future studies should concentrate on improving on existing analytical techniques to address the changing demands of customized medicine and cancer treatments.
Acknowledgement
Authors thankVishnu Institute of Pharmaceutical Education and Research for access to library facilities.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Mattiuzzi, C.; Lippi,G. Epidemiol. Glob. Health2019, 9,217-222.
CrossRef - Bispo, J.A.B.; Pinheiro,P.S.; Kobetz, E.K. Cold Spring Harb. Perspect. Med.2020, 10,a034819.
CrossRef - Ning, L.; Hu, C.; Lu, P.; Que, Y.; Zhu, X.; Li, D. Hematol. Oncol.2020, 9,29.
CrossRef - Conroy, R. Cancer Network2022.
- Rim, M.H.;Karas, B.L.; Barada, F.; Dean, C.;Levitsky, A.M. J. Health-Syst. Pharm.2024,81, 733-738.
CrossRef - Attwa, M.W.; Abdelhameed, A.S.; Kadi,A.A. Medicina (Kaunas)2024, 60(12),1914.
CrossRef - Smyth, L.M.; Tamura, K.; Oliveira, M.;Ciruelos, E.M.; Mayer, I.A.; Sablin, M.P.;Biganzoli, L.; Ambrose,H.J.; Ashton, J.; Barnicle, A.; Cashell, D.D.; Corcoran, C.; de Bruin, E.C.; Foxley, A.; Hauser, J.; Lindemann, J.P.O.; Maudsleyx, J.P.O.; McEwen, R.; Moschetta, M.; Pass, M.; Rowlands, V.; Schiavon,G.;Banerji, U.; Scaltriti, M.; Taylor,B.S.;Chandarlapaty, S.; Baselga, J.; Hyman, D.M. Cancer Res.2020,26(15), 3947-3957.
CrossRef - Smyth, L.M.; Batist,G.;Meric-Bernstam, F.; Kabos, P.; Spanggaard, I.; Lluch, A.; Jhaveri, K.; Barga, A.; Wong, A.; Schram, A.M.; Ambrose, H.; Carr, T.H.; de Bruin, E.C.; Salinas-Souza, C.; Foxley, A.; Hauser, J.; Lindemann, J.P.O.; Maudsley, R.; McEwen, R.; Moschetta, M., Nikolaou, M.; Schiavon, G.; Razavi, P.; Banerji, U.; Baselga, J.; Hyman, D.M.;Chandarlapaty,S. NPJ Breast Cancer2021,7(1),44.
CrossRef - Andrikopoulou, A.; Chatzinikolaou,S.;Panourgias, E.; Kaparelou, M.; Liontos, M.; Dimopoulos, M.A.; Zagouri, F.Breast2022, 63,157-167.
CrossRef - FDA approves capivasertib with fulvestrant for breast cancer, US Food and Drug Administration,2023.
- New Drug Therapy Approvals 2023 (PDF), US Food and Drug Administration (FDA) (Report),2024.
- Gaur,B.; Sahu, S.;Kori,M.L. J. Pharm. Qual. Assur.2024, 15(2), 640-645.
- Yoshito, G.; Takeo, Y. BPB Reports2024, 7(6), 206-210.
CrossRef - Zhe, Z.; Li, N.; Meng-Lei, C.; Ming-Hui, L.; Wen-Wu, C. Chromatogr.2020, 34(10), e4920.
- Keam,S.J.Drugs2023, 83(6), 547-553.
CrossRef - Telaraja, D.; Kasamon, Y.L.; Collazo, J.S.; Leong, R.; Wang, K.; Li, P.;Dahmane, E.; Yang, Y.; Earp, J.; Grimstein, M.; Rodriguez, L.R.; Theoret, M.R.; Gormley, N.J. Cancer Res.2023, 30,OF1-OF6.
CrossRef - S. Food and Drug Administration (FDA), 1 December 2023.
- P.A.R.Jaypirca, European Medicines Agency (EMA), 20 November 2023.
- Aslan, B.; Kismali, G.; Iles, L.R.; Manyam, G.C.; Ayres, M.L.; Chen, L.S.; Gagea,M. Bertilaccio, M.T.S.; Wierda, W.G.; Gandhi,V. Blood Cancer J.2022, 12(5), 80.
CrossRef - Gomez, E.B.; Ebata, K.;Randeria, H.S.; Rosendahl, M.S.; Cedervall, E.P.; Morales,T.H.; Hanson, L.M.; Brown, N.E.; Gong, X.; Stephens, J.; Wu, W.; Lippincott, I.; Ku,K.S.;Walgren, R.A.; Abada, P.B.; Ballard, J.A.;Allerston, C.K.; Brandhuber, B.J.Blood, 2023, 42(1), 62-72.
- Jensen, J.L.;Mato, A.R.; Pena, C.; Roeker, L.E.; Coombs,C.C. Adv. Hematol,2022, 13.
CrossRef - Alu, A.; Lei,H.; Han, X.; Wei, Y.; Wei, X. Hematol. Oncol.2022, 15(1), 138.
CrossRef - Mato, A.R.; Shah, N.N.; Jurczak, W.;Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.;Fakhri,B.; Eyre, T.A.;Lamanna,N.; Patel, M.R.; Alencar, A.; Lech-Maranda, E.; Wierda, W.G.; Coombs, C.C.; Gerson, J.N.; Ghia, P.; Le Gouill, S.; Lewis, D.J.;Sundaram, S.; Cohen,J.B.;Flinn, I.W.; Tam, C.S.; Barve, M.A.; Kuss, B.; Taylor, J.; Abdel-Wahab, O.; Schuster, S.J.; Palomba, M.L.; Lewis, K.L.; Roeker, L.E.; Davids, M.S., Tan, X.N.;Fenske, T.S.; Wallin, J.; Tsai, D.E.; Ku, N.C., Zhu, E.; Chen, J.; Yin, M.; Nair, B.; Ebata,K.; Marella,N.; Brown, J.R.; Wang, M.Lancet 2021, 397,892.
CrossRef - Wang, E.; Mi, X.; Thompson, M.C.; Montoya, S.; Notti, R.Q.; Afaghani, J.; Durham, B.H.; Penson, A.; Witkowski, M.T.; Lu, S.X.;Bourcier, J.; Hogg, S.J., Erickson, C.; Cui,D.; Cho, H.; Singer, M.; Totiger, T.M.; Chaudhry, S., Geyer, M.; Alencar, A.; Linley,A.J.;Palomba, M.L.; Coombs, C.C.; Park, J.H.; Zelenetz, A.; Roeker, L.; Rosendahl,M.; Tsai, D.E.; Ebata, K.; Brandhuber, B.; Hyman, D.M.; Aifantis, I.; Mato, A.; Taylor, J., Abdel-Wahab, O. Engl. J. Med., 2022, 386(8), 735.
CrossRef - Pavithra, M.K.; Chaya, G.; Hemavathi, N.D.; Hosakere,D.R.; Salah Jasim, M.; Hasan,S.M.; Abdullah, H.A.; Shareefraza, J.U. RSC Adv.2024, 14, 34868-34882.
CrossRef - Zhang, M.; Wu, J.; Li, J.; Yin, H.; Hou, M.; Dong,R. BMC Chem.2025, 19(1), 50.
CrossRef - World Health Organization, International Nonproprietary Names for Pharmaceutical Substances (INN), WHO Drug Information 2010, 24(4), 386.
- Jayaraman, R.; Pasha, M.K.; Williams, A.; Goh, K.C.; Ethirajulu, K.Drug Metab.Lett.2015, 9(1), 28-47.
CrossRef - World Health Organization, International Nonproprietary Names for Pharmaceutical Substances (INN): Recommended INN: List 66, WHO Drug Information2011, 25(3), 295-340.
- Phani Kumar, S.; Sreedhara, C.; Ramakrishna, K.J Pharmacol. Toxicol. Methods2024, 129, 107547.
CrossRef - Aruna Kumar, M.; Bangaraiah, P.Separation Science Plus,2024, 7(12), e202400166.
CrossRef - Wu, Y.M.; Huang, L.; Ni, J.H.; Chen, X.; Tang, P.F.; Qian, J.; Xiao, Z.; Xu,H. SSRN2023.
- Griesinger, F.; Curigliano, G.; Thomas, M.; Subbiah, V.; Baik, C.S.; Tan,D.S.W.; Lee,D.H.;Misch, D.; Garralda, E.; Kim, D.W.; van der Wekken, A.J.; Gainor, J.F.; Paz-Ares,L.; Liu, S.V.; Kalemkerian, G.P., Houvras, Y.; Bowles,D.W.; Mansfield, A.S.; Lin, J.J.; Smoljanovic, V.; Rahman, A.; Kong, S.; Zalutskaya, A.; Louie-Gao, M.; Boral, A.L.; Mazières, J. Oncol.2022, 33(11), 1168-1178.
CrossRef - Zhu, Y.; Qin,J.; Wu,W.; Cai,L. Chem.2024,12, 1450692.
CrossRef - Zichen, Z.; Qianlun, P.; Tonglin, S.; Qian, H.; Liping, T.; Ting, F.; Jingyue, K.; Yuhong,C.; Yan, Z.Anti-Cancer Agents Med. Chem.2024,24(11), 867-877.
CrossRef - Rushita, S., Dhirendra Kumar,T.;Sibaji, S. J. All Res.Edu. Scientific Methods,2023, 11(5), 2997-3008.
- Rajesh, V.B.; Pavani, P.; Venkata, S.T.; Battula, S.R. Sci. Technol.2024, 37(5), 280-294.
Accepted on: 02 Apr 2026
Second Review by: Dr. Sumit Kumar
Final Approval by: Dr. Pounraj Thanasekaran







ISSN Online: 2231-5039

![]()




{kind=link}