Selective Cyclisation of 2-(2-Hydroxy-phenyl)-2,3-dihydro-1H-quinazolin-4 one with Formaldehyde and Isolation of Two Isomeric Oxazines
 , Priya and Arif Ali Khan *
, Priya and Arif Ali Khan *University School of Basic and Applied Sciences, Guru Gobind Singh Indraprastha University, Dwarka, New Delhi, India.
Corresponding Author E-mail:arif@ipu.ac.in
DOI : http://dx.doi.org/10.13005/ojc/420204
Download this article as:
![]()
A novel series of oxazine-quinazoline compounds was designed and synthesized via the cyclization of 2-(2-hydroxyphenyl)-2,3-dihydro-1H-quinazolin-4-one (1) with formaldehyde, affording 12,12a-dihydro-5-oxa-6a,12-diaza-benzo[a]anthracen-7-one (2) and 10b,11-dihydro-6-oxa-4b,11-diaza-chrysen-12-one (3) in toluene and tetrahydrofuran, respectively.After the products were isolated, elemental analysis, GC-MS, ¹H and ¹³C NMR spectroscopy were used to confirm their chemical structures. For the synthesised oxazines, DFT calculations and molecular docking have been studied.The synthesised compounds showed better binding affinities toward DNA gyrase (PDB ID: 1KZN) and dihydrofolate reductase (PDB ID: 8SRW) in molecular docking studies, suggesting their potential antibacterial activity. Molecular dynamics (MD) simulations further validated stable protein–ligand interactions, highlighting the structural appropriateness of these molecules for biological applications. Collectively, these novel oxazine–quinazoline hybrids represent promising potential as antibacterial and anticancer agents and require further biological investigation.
KEYWORDS:Antibacterial; Energy gap; HOMO-LUMO interactions; Heterocyclic; Molecular docking; Molecular Dynamic Simulation; Oxazines
Introduction
One of the most developing areas of chemistry is heterocyclic chemistry.Since the structural components of heterocycles are found in many naturally occurring chemicals, including vitamins, hormones and antibiotics, they are abundant in nature and essential to life 1. As a result, they have received considerable attention for synthesising biologically active compounds2. Oxazines are heterocyclic molecules with a broad spectrum of biological activity, including anticancer and antibacterial properties 3–11. Therefore, the biochemical and medicinal properties of 1,3-oxazine moieties have attracted the interest of chemists to synthesize their novel derivatives. Derivatives of quinazoline are a type of condensed heterocycle that exhibit significant antibacterial and anticancer properties 12,13, and these compounds are also known to act as growth inhibitors against leukaemia cells 14. Min Wang et.al 15 reported the formation of 1 as the condensation product of salicylaldehyde and 2-anthranilamide. The cyclisation of this quinazolinone was reported withdichlorophenylphosphinein the vicinity oftriethylamine, resulting in the formation of two isomeric products without any selectivity 16.In continuation of our efforts to explore selective ring-closure reactions of quinazolinone derivatives, we now describe the selective cyclization of compound 1 with formaldehyde. Owing to its high reactivity and versatility as a one-carbon building block, formaldehyde can readily undergo addition reactions with a broad range of substrates, thereby facilitating diverse cyclization and heterocycle-forming transformations. Its efficient incorporation into organic frameworks makes it an attractive reagent for constructing structurally complex and functionally rich heterocyclic systems17.Solvent-based control over reaction conditions, which produces the necessary isomers, is a key strategy for selectivity in chemistry and research18.However, when this cyclisation reaction was tried with formaldehyde in solvents with different polarity it was observed that only one of the two isomeric products was formed selectively in each solvent.
Materials and Methods
Salicylaldehyde and Formaldehyde were procured from CDH, and anthranilamide was acquired from Sigma-Aldrich. Toluene was purchased from SDFCL, and tetrahydrofuran was acquired from Sigma-Aldrich and was purified and stored under nitrogen. TLC was carried out by MERCK TLC Silica gel 60F254 plates and iodine for visualization. The NMR spectra of the sample were analyzed by a JEOL resonance spectrophotometer. The commonly used reference compound for figuring out chemical shifts in both ¹H-NMR and ¹³C-NMR spectroscopy is tetramethylsilane (TMS). Mass spectra were obtained by employing a Shimadzu GC-MS QP2010 Ultra spectrometer.
The procedure for the preparation of compounds
Synthesisof 2-(2-Hydroxy-phenyl)-2,3-dihydro-1H-quinazolin-4-one (1): Compound (1) was synthesized as per methodreported by Min Wang et.al19.
Synthesis of 12,12a-Dihydro-5-oxa-6a,12-diaza-benzo[a]anthracen-7-one (2)
Toluene (20 ml) was used to dissolve compound 1(0.24 g, 1 mmol), and formaldehyde (0.11 ml, 1 mmol) was then appended. After mixing the reaction solutions overnight at room temperature,and then filtered.From TLC, we checked the progress of the reaction (ethyl acetate: hexane,20:80). Reduced pressure was used to evacuate the solvent once the reaction was concluded, and dichloromethane was used to recrystallizecrude solid. A pure white product (2) was obtained. Melting point -204℃ 206℃, Yield-75.3%, 13C-NMR (100 MHz, DMSO-d6):δ161.047,153.218(Ar-C-O-),145.198(Ar-C-NH),133.631,129.357,128.389,126. 770,124.039,120.742,120. 254,118.126,115.894,115.175 (Aromatic region), 75.272(N-CH2-O), 69.369(NH-CH-N) 1H-NMR (400 MHz, DMSO-d6): δ 6.477-7.143(aromatic region),6.028(N-CH2-O-),5.607(N-CH-N),5.246(Ar-NH-C), Mass fragments:77((C6H6)+, 40%),121(C7H7N0,30%), 132((C8H7NO)+,100%),251((C15H12N2O2)+, 25%),252(C15H12N2O2,65%). Calculation for elemental analysis of C15H12N2O2:(C,71.42; H,4.79; N,11.10). Found:( C,71.13; H,4.69; N,11.03).
Synthesis of 10b,11-Dihydro-6-oxa-4b,11-diaza-chrysen-12-one (3)
Tetrahydrofuran (20 ml) was used to dissolve compound 1 (0.24 g, 1 mmol), and formaldehyde (0.11 ml, 1 mmol) was then appended. After mixing the reaction solutions overnight at room temperature,and then filtered. From TLC, we checked the progress of the reaction (ethyl acetate: hexane,20:80). Reduced pressure was used to evacuate the solvent once the reaction was finished, and dichloromethane was used to recrystallize the crude solid. A pure white product (3) was obtained. Melting point-190℃-192℃, Yield-79.3%, 13C-NMR (100 MHz, DMSO-d6): δ 161.728 ,153.151(Ar-C-O-), 144.996(Ar-C-N), 133.545,129.137, 127.872, 126.387, 125.179, 120.551, 120.330, 117.829, 115.941, 114.907(Aromatic region), 75.291(N-CH2-O),64.597(NH-CH-N), 1H-NMR (400 MHz, DMSO-d6): δ 9.032 (CONH), 6.578-7.586(aromatic region),6.071(N-CH2-O-),5.380(N-CH-N-).Mass fragments: 77((C6H6)+,40%),121(C7H7N0,30%), 132((C8H7NO) +,100%),
251((C15H12N2O2) +, 25%), 252(C15H12N2O2, 60%). Calculation for elemental analysis of C15H12N2O2🙁 C,71.42; H,4.79; N,11.10). Found: (C,71.15; H,4.62; N,11.06).
Computational analysis
ADMET prediction studies
The ADME prediction was done online by Swiss ADME (http://www.swissadme.ch/)20.Theanalysis provided insights into key pharmacokinetic parameters, including gastrointestinal (GI)absorption, molecular weight, hydrogen-bonding capacity (number of donors and acceptors), compliance withLipinski’s Rule of Five and lipophilicity as indicated by MolLogP.The toxicological prediction was done with ProTox III (https://tox.charite.de/protox3/) server 21.
Molecular docking
Molecular docking is often utilized for antibacterial screening to determine the correct binding mode of chemical compounds that have been tested within the desired protein’s active site.
Protein preparation
The Protein Database (PDB) (https://www.rcsb.org/) supplied the 3D crystal structures of the proteins DNA Gyrase (E. coli) (PDB ID: 1KZN) and Dihydrofolate Reductase (DHFR) (PDB ID: 8SRW) in.pdb format. (retrieved January 27, 2025). The PDB is a global repository for the 3D structures of biological macromolecules22. Heteroatoms, default ligands, and water molecules were eliminated in order to prepare the receptors. The processed receptors were further prepared for docking in AutoDock (version 1.5.6) by adding Kollman charges and polar hydrogen atoms, and then converted to PDBQT format.
Preparation of ligands
The 2D formation of prepared compounds were sketched in ChemDraw Ultra 7.0 .cdx. The ChemDraw structures files were then converted into .pdbqt format using AutoDock.
Molecular docking
Discovery Studio 2021 was utilized to determine the inhibitor binding site based on the active sites found in the PDB site information. The grid and origin dimensions (x, y, and z) encompass all of the traces of amino acids that take place at the active location. The grid points were kept 0.375 Å apart. AutoDock Vina (version 1.5.6) was employed to dock all the ligands to the corresponding proteins’ active sites23.To ensure Vina produced the best findings in a reasonable period of time, the docking exhaustiveness was maintained at 20 throughout this in-silico analysis. The complexes of protein and ligand were displayed employing the Discovery Studio Visualizer (version 21.1.0.20298).
Molecular Dynamics Simulation
GROMACS 2024.2 software was used to select materials that have both protein targets for MD simulations24. TheCHARMM27 was chosen as an all-atom force field25, and SwissParam server26 determined the topology of proteins and ligands26. The complex was stabilized utilizing parameters such as temperature (T), no. of particles (N), isobaric-isothermic ensemble system volume (V), and NVT following it was solved using the TIP3P water model and treated with ions (Cl –,Na+) for 100 picoseconds .At standard temperature and pressure, the NPT group conducted an additional Molecular Dynamics run of complexes of protein-ligand for 100 nanoseconds. The obtained data was utilized to determine the root mean square deviation , the root mean square fluctuations for the protein and ligand, and the radius of gyration of the ligand in the complex in order to comprehend the appropriate dynamic changes of the protein–ligand combination at the molecular level.
Density Functional Theory
The GaussView software has been used to generate the initial structures of the products. Density functional theory (DFT) computations are being carried out utilizing the B3LYP hybrid functional with 6–311 + (d,p) basis sets and Gaussian16 program.27,28. The optimisation of the products’ geometry was performed in the gas phase. The energy gap arising from the values of the LUMO (lowest unoccupied molecular orbital) and HOMO (highest occupied molecular orbital) may be computed using the frontier molecular orbital theory, which was fully described by Parr and Pearson. The ionisation potential (I) and electron affinity (A) are linked to the energy levels of HOMO and LUMO because they have a tendency to donate and acquire electrons, respectively. In order to describe the overall reactivity and stability of a molecular system, some examples of global reactivity descriptors incorporatechemical potential,global hardness, electrophilicity, global softness ,nucleophilicity symbolised as µ,η,ω,S and N respectively. The following is an equation for the global reactivity descriptors:
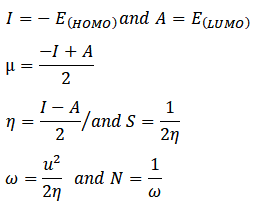
Results and Discussion
Characterisation of synthesized compounds
The compound 1 shows the difficulty in the isolation of two rotomers in pure form because these rotomers are distinguished by a small amount of energy. However, when this compound was cyclized with formaldehyde in the presence of an appropriate solvent, the product of two rotomers was formed. It is a phenomenal outcome of the parent compound that rotates around the C-C single bond. The compound 1consists of a quinazolinone ring and a phenyl ring joined by a C-C bond. The phenyl ring with the hydroxyl group is free to rotate along the C-C axis. One of the 2 (-NH) groups on the quinazolin-4-one ring at a time can cyclise with the hydroxyl group in the presence of formaldehyde. TLC monitored the reaction progress. The two compounds (2 and 3) were formed with distinct melting points and solubilities (toluene and tetrahydrofuran), respectively. In GC-MS, these two compounds have different retention times but the same molecular mass (252(base peak), R. time: 20.303 min, R. time: 20.233 min), respectively. Additionally, 13C NMR and 1H NMR can be used to differentiate them. Compounds (2) and (3) were recrystallised using dichloromethane.
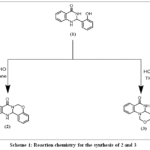 |
Scheme 1: Reaction chemistry for the synthesis of 2 and 3 Click here to View Scheme |
HOMO-LUMO study
The frontier molecular orbital (FMO) explains the HOMO-LUMO relationship among molecules, as well as a molecule’s inclination to transfer electrons to handy molecules with low-energy empty molecular orbitals 30. HOMO exhibits nucleophilic behaviour (the capacity to donate electrons) while LUMO exhibits electrophilic behaviour (the capacity to accept electrons). Fig. 1 displays the HOMO-LUMO interaction of their orbitals and their energy gaps of the compounds, which explains the compounds’ thermodynamic stability and chemical reactivity. A smaller energy gap(ΔE) typically indicates greater chemical reactivity, which results in improved compound-enzyme interaction and enhances their antibacterial potential¹. Lower ΔE values indicate enhanced polarizability and electronic softness, which increase electron transfer and binding interactions with bacterial enzymes or cell-wall targets31.A smaller gap typically indicates greater chemical reactivity, which could result in improved compound-enzyme interaction. The small energy gap of the compounds may allow them to have antibacterial properties. Compounds’ capacity to donate and accept electrons chemical potential, hardness, and softness is described by global reactivity descriptions.
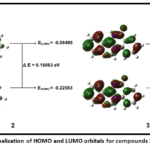 |
Figure 1: Visualization of HOMO and LUMO orbitals for compounds 2 and 3. Click here to View Figure |
Table 1: Illustration ofglobal reactivity descriptors for compounds 2 and 3.
|
Symbol |
Property | 2 | 3 |
| I | Ionization energy | 0.22553 |
0.06305 |
|
A |
Electron affinity | 0.05499 | 0.14438 |
| μ | Chemical potential | -0.14026 |
-0.103715 |
|
Ƞ |
Global hardness | 0.08527 | 0.04066 |
| S | Global softness | 5.8637 |
12.295 |
|
ω |
Electrophilicity | 0.11535 | 0.13226 |
| N | Nucleophilicity | 8.66926 |
7.5608 |
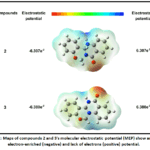 |
Figure 2: Maps of compounds 2 and 3’s molecular electrostatic potential (MEP) show areas of electron-enriched (negative) and lack of electrons (positive) potential. Click here to View Figure |
Molecular electrostatic potential (MEP) analysis
Finding and evaluating the chemical’s areas of electrophile assault that are relatively reactive, hydrogen bonding, and nucleophile attack 32 is a pretty simple process. The chk file of the optimized structures was used to forecast the 3D MEP plot of both compounds.Electrophilic reactivity and negative electrostatic potential (oxygen group) are indicated by the color red when the proton is attracted, nucleophilic reactivity and positive electrostatic potential (nitrogen group) are indicated by the color blue when the proton is repelled, and zero electrostatic potential is indicated by the color green33. A potent red area may be close to a carbonyl group that may create powerful H-bonds with bacterial enzyme active sites, while a blue area indicates that it might interact with bacterial cell walls and nucleic acids. The electrostatic potential of both compounds 2 and 3 falls between -6.307e -2 to 6.307e 2 and -6.380e -2 to 6.380e 2, respectively.
ADME and toxicity prediction studies
In drug development and research, clinical trials are not considered for potential drug candidates with unfavorable (ADME) criteria. According to Table 2, the generated compounds satisfied all five standards of Lipinski’s principle of five.No violation indicated that the candidates may work well as oral medications. Additionally, certain substances demonstrated high bioavailability and gastrointestinal (GI) absorption ratings (0.55) and were predicted to be absorbed orally. Additionally, the ProTox3 server’s estimated Median Lethal Dose (LD50) values in mg/Kg were used to analyze the toxicity characteristics of a few chosen substances. Both synthesized compounds had LD50 values of 1500 mg/kg, which indicated reduced toxicity.Significantly, these pharmacokinetic properties and toxicity results show that the synthesized compounds have desirable characteristics of drug-like molecules, providing a strong rationale for their potential antibacterial and antimicrobial potential and demand further biological testing.
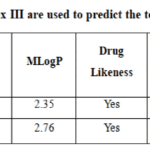 |
Table 2: SwissADME and ProTox III are used to predict the toxicity and ADME of substances. Click here to View Table |
Molecular Docking Studies
The antibacterial property of the created oxazines 2 and 3 was postulated utilizing molecular docking screening. To investigate the antibacterial activity, the E. Coli DNA gyrase enzyme (PDB ID: 1KZN) and S. aureus DHFR (PDB ID: 3SRW) were selected as possible therapeutic targets. The DNA Gyrase enzyme is one of the topoisomerases (topoisomerase II). During transcription and replication, these enzymes wind and unwind the DNA. The gyrase enzyme is anticipated to be a crucial intracellular target for antibacterial drugs34 due to its capacity to alter the topological state of DNA and serve as a model for other DNA topoisomerases.An enzyme called DHFR is essential to the metabolic processes of all cells, including those of bacteria and humans. Its main job is to use NADPH as a cofactor to catalyse the reduction of dihydrofolate (DHF) into tetrahydrofolate (THF). Tetrahydrofolate is a necessary cofactor for the production of certain amino acids and nucleotides, which are the building blocks of Protein, DNA and RNA, respectively. As long as the medications are selective, bacterial DHFR inhibitors can efficiently eradicate or stop bacterial development without seriously damaging human cells35.
Molecular Docking analysis showed that synthesised compounds 2 and 3 showed better binding affinity with 3SRW and form Hydrogen bonds, other than vanderwaal forces, alkyl and pi-alkyl bonds. While with 1KZN vanderwaal forces, alkyl and pi-alkyl bonds are the only interactions observed.
Table 3: Molecular docking results showing the binding affinities of the synthesized compounds toward 1KZN and 3SRW.
| Ligand |
Binding Affinity 1KZN (kcal/mol) |
Interacting
H-bond Residue |
Binding Affinity
3SRW (kcal/mol) |
Interacting H-bond Residue |
| 2 | -8.0 | – | -9.5 | SER50 |
| 3 | -8.2 | – | -9.0 | SER50, THR47 |
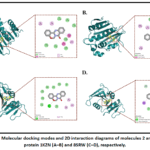 |
Figure 3: Molecular docking modes and 2D interaction diagrams of molecules 2 and 3 with protein 1KZN (A–B) and 8SRW (C–D), respectively. Click here to View Figure |
Molecular dynamics simulation
The dynamic image of the biological system was anticipated using MD modelling. It took into account a number of biological elements, including co-enzymes, ions, and the surrounding environment, including water. It operated by offering a perfect model that included every physical circumstance that was utilised to explain a biological system’s dynamics. The formation of hydrogen bonds is necessary for the consistency of shape, catalytic region, protein–ligand interaction, and structural integrity of the protein.The hydrogen bond interaction within the protein changes in antibacterial activity for all complexes during the 100 ns MD simulation, as seen in Figures 4G and 4H. Compared to 1KZN, the hydrogen bond values of proteins, complex 2, and complex 3 with 8SRW were higher.
We are able to assess the variation across residues by using RMSF36. As can be seen, all of the antibacterial complexes showed comparable fluctuations to those found in protein structures (Figure 4E & 4F), suggesting that the complexes were significantly more stable and had lower energy than the protein close to certain residuesThe fundamental structure of intricate antimicrobial proteins was seen using the RMSD technique. While the protein complexes for ligand 2 and 3 demonstrated more stability and lower RMSD values for the 8SRW protein, all of the complexes had stabilized patterns with almost identical RMSD values ranging between 0.2 and 0.3 nm (Figure 4A & 4B). The backbone deviation of a protein can be inferred from the RMSD fluctuations37. Variations in the ligand-protein complex’s compactness are measured by the radius of gyration (Rg). Additionally, Rg controls how proteins and their complexes fold and unfold steadily. A high Rg indicates a less compact protein-ligand interaction. Protein folding is stable when Rg remains relatively constant throughout MD modeling; if Rg fluctuates over time, the protein is unraveling38. The Rg values of the protein and protein-ligand complex were comparatively constant, and the protein-ligand complexes for the 8SRW protein exhibited a similar pattern. As a result, the complexes of both ligand 2 and 3 with the proteins 1KZN and 8SRW provided excellent dynamic structural stability while restricting the protein’s flexibility.
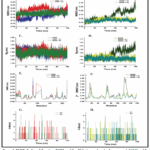 |
Figure 4: RMSD, Radius of Gyration, RMSF, and No. Of Hydrogen bonds of compounds 2 (A1) and 3 (A2) complex with 1KZN and 8SRW protein after 100 ns of MD simulation. Click here to View Figure |
Conclusion
The two isomeric products are very interesting compounds as each one has an oxazine ring in it and may have potential applications as antitumor and antimicrobial reagents. Formaldehyde, along with an appropriate solvent could be used to selectively protect either of the N-H bonds, and the substitution of another N-H bond with suitable groups may result in the isolation of novel compounds. The antibacterial potential of these compounds was predicted using a molecular docking approach, which can be further validated using in vitro and in vivo studies. Through the cyclization of 1 with formaldehyde, the current study successfully synthesized novel oxazine–quinazoline hybrids, producing two structurally verified derivatives in the solvents tetrahydrofuran and toluene. Their chemical identities were determined through thorough characterisation utilizing GC-MS, NMR, and elemental analysis. According to molecular docking studies, these substances have higher binding affinities for two important antibacterial targets: bacterial DNA gyrase and dihydrofolate reductase. Stable and advantageous protein–ligand interactions were further validated by molecular dynamics simulations, confirming their structural appropriateness for biological applications.
Acknowledgment
We are grateful to the University School of Basic and Applied Sciences for the technical support.
Funding Sources
This research was funded by GGSIP University for providing a Short-Term Research Fellowship (STRF) and a FRGS Grant.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Author Contribution’s
- Gourav Arora: Writing – original draft, Synthesis, Isolation and Collection of analytical data.
- Priya: Writing – review & editing, Software.
- Arif Ali Khan: Writing – original draft, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
References
- Ju, Y.; Varma, R. S. Aqueous N-Heterocyclization of Primary Amines and Hydrazines with Dihalides: Microwave-Assisted Syntheses of N-Azacycloalkanes, Isoindole, Pyrazole, Pyrazolidine, and Phthalazine Derivatives. J. Org. Chem.2006, 71 (1), 135–141. https://doi.org/10.1021/jo051878h.
CrossRef - Nagaraj, A.; Sanjeeva Reddy, C. Synthesis and Biological Study of Novel Bis-Chalcones, Bis-Thiazines and Bis-Pyrimidines. J. Iran. Chem. Soc.2008, 5 (2), 262–267. https://doi.org/10.1007/BF03246116.
CrossRef - Ouberai, M.; Asche, C.; Carrez, D.; Croisy, A.; Dumy, P.; Demeunynck, M. 3,4-Dihydro-1H-[1,3]Oxazino[4,5-c]Acridines as a New Family of Cytotoxic Drugs. Bioorg. Med. Chem. Lett.2006, 16 (17), 4641–4643. https://doi.org/10.1016/j.bmcl.2006.05.101.
CrossRef - Mathew, B. P.; Kumar, A.; Sharma, S.; Shukla, P. K.; Nath, M. An Eco-Friendly Synthesis and Antimicrobial Activities of Dihydro-2H-Benzo- and Naphtho-1,3-Oxazine Derivatives. Eur. J. Med. Chem.2010, 45 (4), 1502–1507. https://doi.org/10.1016/j.ejmech.2009.12.058.
CrossRef - Tang, Z.; Zhu, Z.; Xia, Z.; Liu, H.; Chen, J.; Xiao, W.; Ou, X. Synthesis and Fungicidal Activity of Novel 2,3-Disubstituted-1,3-Benzoxazines. Molecules2012, 17 (7), 8174–8185. https://doi.org/10.3390/molecules17078174.
CrossRef - Sawant, R.; Mhaske, M. S.; Wadekar, J. Anticoagulant potential of schiff bases of 1, 3-oxazines. Int. J. PharmTech Res,2012, 4, 320-323.
- Henry, B. L.; Desai, U. R. Recent Research Developments in the Direct Inhibition of Coagulation Proteinases – Inhibitors of the Initiation Phase. Cardiovasc. Hematol. Agents Med. Chem. Former. Curr. Med. Chem. – Cardiovasc. Hematol. Agents2008, 6 (4), 323–336. https://doi.org/10.2174/187152508785909519.
CrossRef - Thompson, A. M.; Blaser, A.; Anderson, R. F.; Shinde, S. S.; Franzblau, S. G.; Ma, Z.; Denny, W. A.; Palmer, B. D. Synthesis, Reduction Potentials, and Antitubercular Activity of Ring A/B Analogues of the Bioreductive Drug (6S)-2-Nitro-6-{[4-(Trifluoromethoxy)Benzyl]Oxy}-6,7-Dihydro-5H-Imidazo[2,1-b][1,3]Oxazine (PA-824). J. Med. Chem.2009, 52 (3), 637–645. https://doi.org/10.1021/jm801087e.
CrossRef - Anderson, R. F.; Shinde, S. S.; Maroz, A.; Boyd, M.; Palmer, B. D.; Denny, W. A. Intermediates in the Reduction of the Antituberculosis Drug PA-824, (6S)-2-Nitro-6-{[4-(Trifluoromethoxy)Benzyl]Oxy}-6,7-Dihydro-5H-Imidazo[2,1-b][1,3]Oxazine, in Aqueous Solution. Org. Biomol. Chem.2008, 6 (11), 1973–1980. https://doi.org/10.1039/B801859F.
CrossRef - Benaamane, N.; Nedjar-Kolli, B.; Bentarzi, Y.; Hammal, L.; Geronikaki, A.; Eleftheriou, P.; Lagunin, A. Synthesis and in Silico Biological Activity Evaluation of New N-Substituted Pyrazolo-Oxazin-2-One Systems. Bioorg. Med. Chem.2008, 16 (6), 3059–3066. https://doi.org/10.1016/j.bmc.2007.12.033.
CrossRef - Madhavan, G. R.; Chakrabarti, R.; Anantha Reddy, K.; Rajesh, B. M.; Balraju, V.; Bheema Rao, P.; Rajagopalan, R.; Iqbal, J. Dual PPAR-α and -γ Activators Derived from Novel Benzoxazinone Containing Thiazolidinediones Having Antidiabetic and Hypolipidemic Potential. Bioorg. Med. Chem.2006, 14 (2), 584–591. https://doi.org/10.1016/j.bmc.2005.08.043.
CrossRef - Parish, H. A. Jr.; Gilliom, R. D.; Purcell, W. P.; Browne, R. K.; Spirk, R. F.; White, H. D. Syntheses and Diuretic Activity of 1,2-Dihydro-2-(3-Pyridyl)-3H-Pyrido[2,3-d]Pyrimidin-4-One and Related Compounds. J. Med. Chem.1982, 25 (1), 98–102. https://doi.org/10.1021/jm00343a022.
CrossRef - Alaimo, R. J.; Russell, H. E. Antibacterial 2,3-Dihydro-2-(5-Nitro-2-Thienyl)Quinazolin-4-(1H)-Ones. J. Med. Chem.1972, 15 (3), 335–336. https://doi.org/10.1021/jm00273a034.
CrossRef - Li, X.; Manjunatha, U. H.; Goodwin, M. B.; Knox, J. E.; Lipinski, C. A.; Keller, T. H.; Barry, C. E.; Dowd, C. S. Synthesis and Antitubercular Activity of 7-(R)- and 7-(S)-Methyl-2-Nitro-6-(S)-(4-(Trifluoromethoxy)Benzyloxy)-6,7-Dihydro-5H-Imidazo[2,1-b][1,3]Oxazines, Analogues of PA-824. Bioorg. Med. Chem. Lett.2008, 18 (7), 2256–2262. https://doi.org/10.1016/j.bmcl.2008.03.011.
CrossRef - Wang, M.; Zhang, T. T.; Song, Z. G. Eco-Friendly Synthesis of 2-Substituted-2,3-Dihydro-4(1H)-Quinazolinones in Water. Chin. Chem. Lett.2011, 22 (4), 427–430. https://doi.org/10.1016/j.cclet.2010.10.038.
CrossRef - Neetu, Khan, A. A. Freezing the Rotation around Carbon-Carbon Bond in Two Inseparable Conformers by Cyclisation and Isolation of Their Products. Org. Med. Chem. Int. J. 2021,11,555810.
- Attorresi, C. I.; Ramírez, J. A.; Westermann, B. Formaldehyde Surrogates in Multicomponent Reactions. Beilstein J. Org. Chem.2025, 21 (1), 564–595. https://doi.org/10.3762/bjoc.21.45.
CrossRef - Meng JP, Wang WW, Chen YL, Bera S, Wu J. Switchable solvent-controlled divergent synthesis: an efficient and regioselective approach to pyrimidine and dibenzo [b, f][1, 4] oxazepine derivatives. Organic Chemistry Frontiers. 2020;7(2):267-72.
CrossRef - Wang, S.; Li, Y.; Liu, Y.; Lu, A.; You, Q. Novel Hexacyclic Camptothecin Derivatives. Part 1: Synthesis and Cytotoxicity of Camptothecins with an A-Ring Fused 1,3-Oxazine Ring. Bioorg. Med. Chem. Lett.2008, 18 (14), 4095–4097. https://doi.org/10.1016/j.bmcl.2008.05.103.
CrossRef - Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep.2017, 7, 42717. https://doi.org/10.1038/srep42717.
CrossRef - Banerjee, P.; Kemmler, E.; Dunkel, M.; Preissner, R. ProTox 3.0: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res.2024, 52 (W1), W513–W520. https://doi.org/10.1093/nar/gkae303.
CrossRef - Berman, H. M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T. N.; Weissig, H.; Shindyalov, I. N.; Bourne, P. E. The Protein Data Bank. Nucleic Acids Res.2000, 28 (1), 235–242. https://doi.org/10.1093/nar/28.1.235.
CrossRef - Trott, O.; Olson, A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem.2010, 31 (2), 455–461. https://doi.org/10.1002/jcc.21334.
CrossRef - Abraham, M. J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J. C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX2015, 1–2, 19–25. https://doi.org/10.1016/j.softx.2015.06.001.
CrossRef - Bjelkmar, P.; Larsson, P.; Cuendet, M.; Lindahl, E. Implementation of the CHARMM Force Field in GROMACS : Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput.2010, 6 (2), 459–466.
CrossRef - Zoete, V.; Cuendet, M. A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem.2011, 32 (11), 2359–2368. https://doi.org/10.1002/jcc.21816.
CrossRef - Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H. Gaussian 16 Revision C. 01, 2016. Gaussian Inc Wallingford CT2016, 1, 572.
- Kącka-Zych, A.; Pérez, P. Perfluorobicyclo[2.2.0]Hex-1(4)-Ene as Unique Partner for Diels–Alder Reactions with Benzene: A Density Functional Theory Study. Theor. Chem. Acc.2021, 140 (2), 17. https://doi.org/10.1007/s00214-020-02709-6.
CrossRef - Domingo, L. R.; Ríos‐Gutiérrez, M. Application of Reactivity Indices in the Study of Polar D Iels– A Lder Reactions. In Conceptual Density Functional Theory; Liu, S., Ed.; Wiley, 2022; pp 481–502. https://doi.org/10.1002/9783527829941.ch24.
CrossRef - Honda, S.; Fukuyama, Y.; Nishiwaki, H.; Masuda, A.; Masuda, T. Conversion to Purpurogallin, a Key Step in the Mechanism of the Potent Xanthine Oxidase Inhibitory Activity of Pyrogallol. Free Radic. Biol. Med.2017, 106, 228–235. https://doi.org/10.1016/j.freeradbiomed.2017.02.037.
CrossRef - Zinad, D. S.; Mahal, A.; Salman, G. A.; Shareef, O. A.; Pratama, M. R. F. Molecular Docking and DFT Study of Synthesized Oxazine Derivatives. Egypt. J. Chem.2022, 65 (7), 231–240. https://doi.org/10.21608/ejchem.2021.102664.4755.
CrossRef - Abuelizz, H. A.; Taie, H. A. A.; Bakheit, A. H.; Marzouk, M.; Abdellatif, M. M.; Al-Salahi, R. Biological Evaluation of 4-(1H-Triazol-1-Yl)Benzoic Acid Hybrids as Antioxidant Agents: In Vitro Screening and DFT Study. Appl. Sci.2021, 11 (24), 11642. https://doi.org/10.3390/app112411642.
CrossRef - Luque, F. J.; Orozco, M.; Bhadane, P. K.; Gadre, S. R. SCRF Calculation of the Effect of Water on the Topology of the Molecular Electrostatic Potential. J. Phys. Chem.1993, 97 (37), 9380–9384. https://doi.org/10.1021/j100139a021.
CrossRef - Hissam, M. A.; Ngaini, Z.; Zamakshshari, N. H.; Hejemi, F. N. A. M.; Arni, F. S.; Halim, A. N. A. Synthesis and Molecular Docking Simulation on the Antimicrobial Effects of Halogenated Vanillin-Azo Dyes and Schiff Base Derivatives. Discov. Appl. Sci.2024, 6 (6), 325. https://doi.org/10.1007/s42452-024-05830-4.
CrossRef - He, J.; Qiao, W.; An, Q.; Yang, T.; Luo, Y. Dihydrofolate Reductase Inhibitors for Use as Antimicrobial Agents. Eur. J. Med. Chem.2020, 195, 112268. https://doi.org/10.1016/j.ejmech.2020.112268.
CrossRef - Kashyap, J.; Datta, D. Drug Repurposing for SARS-CoV-2: A High-Throughput Molecular Docking, Molecular Dynamics, Machine Learning, and DFT Study. J. Mater. Sci.2022, 57 (23), 10780–10802. https://doi.org/10.1007/s10853-022-07195-8.
CrossRef - Evaluating the Binding Potential and Stability of Drug-like Compounds with the Monkeypox Virus VP39 Protein Using Molecular Dynamics Simulations and Free Energy Analysis. https://www.mdpi.com/1424-8247/17/12/1617 (accessed 2025-09-10).
CrossRef - Lobanov, M. Yu.; Bogatyreva, N. S.; Galzitskaya, O. V. Radius of Gyration as an Indicator of Protein Structure Compactness. Mol. Biol.2008, 42 (4), 623–628. https://doi.org/10.1134/S0026893308040195.
CrossRef
Accepted on: 28 Feb 2026
Second Review by: Dr. K. K.Ilavenil Chemistry
Final Approval by: Dr. Ioana Stanciu







ISSN Online: 2231-5039

![]()




{kind=link}