Design and in Vitro Neuroprotective Evaluation of Pyrimidine-Based Compounds for Alzheimer’s Therapy
 , Gunjan Rani2, Sadhana Rai3, Ayesha Yasmeen4, Mohit Kumar5, Oleti Navneetha6, Sushil Kumar Gulia7, Renu Solanki8and P. Geetha9*
, Gunjan Rani2, Sadhana Rai3, Ayesha Yasmeen4, Mohit Kumar5, Oleti Navneetha6, Sushil Kumar Gulia7, Renu Solanki8and P. Geetha9*
1Goenka College of Pharmacy, Ghassu, lachhmangarh, Sikar, Rajasthan, India.
2School of Pharmacy Mangalayatan University Aligarh Uttar Pradesh, india.
3The ICFAI University Raipur, Chhattisgarh, India.
4Department of Clinical Practice, College of Pharmacy, Jazan University, Jazan, Saudi Arabia
5Teerthanker Mahaveer College of Pharmacy, Teerthanker Mahaveer University; Moradabad, Uttar Pradesh, India.
6Joginpally B. R. Pharmacy College, Yenkapally, Moinabad, Ranga Reddy, Telangana state, India.
7SGT College of Pharmacy, Chandu-Bhudera Gurugram, Haryana, India.
8Department of Quality Assurance, Lachoo Memorial College of Science and Technology (Autonomous) Pharmacy Jodhpur Rajasthan, Sector A Shastri Nagar Jodhpur Rajasthan, India.
9Faculty of Pharmacy, Sree Balaji Medical College and Hospital campus, Bharath Institute of Higher Education and Research, (DU), Chromepet, Chennai, Tamil Nadu, India.
Corresponding Author E-mail: lgeethapharma@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/420212
Download this article as:
![]()
Alzheimer's disease (AD), the leading cause of dementia, presents a complex, multifactorial pathology involving cholinergic deficit, amyloid-beta aggregation, tau hyperphosphorylation, oxidative stress, and metal dyshomeostasis. The repeated failure of single-target therapeutics underscores the urgent need for innovative strategies. The multi-target-directed ligand (MTDL) approach has emerged as a promising paradigm, aiming to design single molecules capable of simultaneously modulating multiple pathological pathways. This review highlights the pyrimidine scaffold as a privileged structure in medicinal chemistry for the development of such neuroprotective agents. Its inherent structural versatility, synthetic accessibility, and favorable drug-like properties allow for strategic substitution to create compounds with potent and balanced biological activities. We detail the evolution from single-target to multi-target design, focusing on hybrid molecules and metal complexation strategies, including the synthesis of pyrimidine-based Schiff bases, thiourea derivatives, hydrazones, and phthalocyanines. A critical analysis of in vitro evaluation methodologies, particularly cholinesterase inhibition assays, is provided, supported by case studies of pyrimidinylthioureas, Schiff base phthalocyanines, and hydrazone derivatives that demonstrate potent and selective acetylcholinesterase and butyrylcholinesterase inhibition alongside antioxidant and metal-chelating properties. The integration of pharmacophore modeling and an understanding of structure-activity relationships is emphasized as essential for optimizing blood-brain barrier permeability and achieving the desired multi-target profile. In conclusion, the rational design of pyrimidine-based MTDLs represents a viable and powerful strategy to combat the complex pathophysiology of Alzheimer's disease, offering a pathway toward more effective disease-modifying therapies.
KEYWORDS:Alzheimer's Disease; Blood-Brain Barrier; Cholinesterase Inhibitors; Hydrazones; Metal Chelation; Neuroprotection; Pyrimidine; Phthalocyanines; Schiff Bases
Introduction
Alzheimer’s disease stands as the most prevalent neurodegenerative disorder and the leading cause of dementia worldwide, representing one of the most significant public health crises of the twenty-first century[1]. This progressive and irreversible brain disorder slowly destroys cognitive function, including memory, thinking skills, and the ability to perform simple tasks[2][3]. The pathological landscape of Alzheimer’s disease is complex and multifactorial, characterized by several interconnected hallmarks that collectively contribute to neuronal dysfunction and death. The two primary pathological features, first described by Alois Alzheimer in 1906, are the extracellular accumulation of amyloid-beta peptides into senile plaques and the intracellular aggregation of hyperphosphorylated tau protein forming neurofibrillary tangles[4]. The amyloid cascade hypothesis has long dominated the understanding of disease pathogenesis, proposing that the aberrant production and impaired clearance of amyloid-beta peptides, particularly the more aggregation-prone Aβ42 isoform, trigger a cascade of events including synaptic dysfunction, neuroinflammation, oxidative stress, and ultimately neuronal loss. However, the repeated failure of clinical trials targeting amyloid-beta alone has highlighted the inadequacy of this unidimensional view and underscored the disease’s true complexity. Beyond these classic proteinopathies[5][6][7], Alzheimer’s pathology encompasses chronic neuroinflammation mediated by activated microglia and astrocytes, profound oxidative stress resulting from mitochondrial dysfunction and metal dyshomeostasis, cholinergic deficit characterized by the degeneration of basal forebrain cholinergic neurons, and widespread synaptic and neuronal loss[8][9]. The interplay between these pathological processes creates a self-perpetuating cycle of neurodegeneration that has proven extraordinarily difficult to interrupt with single-target therapeutic interventions. Furthermore, the diagnosis typically occurs years after pathological changes have begun, meaning that therapeutic interventions often face the challenge of reversing established pathology rather than preventing its initiation. The heterogeneity of disease presentation, the influence of genetic risk factors such as APOE4 genotype, and the complexity of the blood-brain barrier limiting drug access to the central nervous system compound these therapeutic challenges, creating an urgent need for innovative approaches that address the multifactorial nature of Alzheimer’s disease[10][11].
Current Treatment Strategies and Their Limitations
The current pharmacological arsenal against Alzheimer’s disease remains woefully inadequate, offering only modest symptomatic relief without the ability to halt or reverse disease progression[12][13]. The approved therapeutic agents fall into two main categories: the cholinesterase inhibitors and the N-methyl-D-aspartate receptor antagonist memantine. The cholinesterase inhibitors, including donepezil, rivastigmine, and galantamine, operate on the cholinergic hypothesis by preventing the breakdown of acetylcholine in the synaptic cleft, thereby temporarily enhancing cholinergic neurotransmission in brain regions affected by neurodegeneration. While these drugs provide modest cognitive and functional benefits for some patients, their effects are symptomatic and transient, typically lasting six to twelve months before cognitive decline resumes its inexorable trajectory[14][15]. Moreover, their use is limited by peripheral cholinergic side effects including nausea, vomiting, diarrhea, and bradycardia, which can significantly impact patient quality of life and treatment adherence. Memantine, an uncompetitive antagonist of NMDA receptors, works by modulating glutamatergic excitotoxicity, offering additional symptomatic benefit in moderate to advanced disease stages. However, like the cholinesterase inhibitors, memantine does not modify the underlying disease process. The recent conditional approval of aducanumab and the full approval of lecanemab, monoclonal antibodies targeting amyloid-beta, represent a paradigm shift toward disease-modifying therapies, yet their clinical benefits remain modest while their administration requires intravenous infusion and carries risks of amyloid-related imaging abnormalities including cerebral edema and microhemorrhages[16][17]. These high costs, limited accessibility, and significant safety concerns restrict their widespread applicability. The failure of numerous clinical trials targeting various aspects of Alzheimer’s pathology, from secretase inhibitors to anti-inflammatory agents and antioxidants, underscores the difficulty of translating preclinical findings into effective clinical treatments. This therapeutic stagnation reflects the fundamental challenge that Alzheimer’s disease is not a simple linear cascade but a complex network of interacting pathological processes that may require simultaneous modulation of multiple targets for meaningful therapeutic benefit[18].
The Multi-Target-Directed Ligand (MTDL) Approach in AD Drug Discovery
In response to the limitations of single-target therapies and the recognized complexity of Alzheimer’s pathophysiology, the multi-target-directed ligand approach has emerged as a particularly attractive and rational drug discovery paradigm[19][20]. This strategy recognizes that Alzheimer’s disease, like many complex neurodegenerative disorders, involves multiple interconnected pathological pathways that collectively drive disease progression, and that therapeutic agents capable of simultaneously modulating several of these targets may offer superior efficacy compared to combination therapies with multiple single-target drugs[21][22]. The multi-target-directed ligand concept involves the intentional design of a single molecular entity that can interact with two or more complementary biological targets implicated in disease pathogenesis, thereby addressing the network nature of the pathology while potentially avoiding the pharmacokinetic complexities and drug-drug interaction concerns associated with multi-drug cocktails[23][24][25].
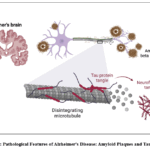 |
Figure 1: Pathological Features of Alzheimer’s Disease: Amyloid Plaques and Tau Tangles Click here to View Figure |
In the context of Alzheimer’s disease, an ideal multi-target-directed ligand might combine cholinesterase inhibition to address the cholinergic deficit, antioxidant activity to counter oxidative stress, metal chelation to normalize metal dyshomeostasis, anti-amyloid aggregation properties to prevent plaque formation, and anti-neuroinflammatory effects to modulate the glial response, all within a single molecular framework[27][28]. The challenge lies in achieving balanced activities against multiple targets without compromising drug-like properties including blood-brain barrier permeability, metabolic stability, and safety profile. The pyrimidine scaffold has emerged as an exceptionally promising platform for multi-target-directed ligand development due to its inherent structural flexibility, synthetic accessibility, and demonstrated ability to interact with diverse biological targets through appropriate substitution patterns. By strategically modifying the pyrimidine core with various functional groups, medicinal chemists can create molecules that engage with cholinesterase enzymes, chelate redox-active metals, scavenge free radicals, and modulate protein aggregation, all while maintaining the physicochemical properties necessary for central nervous system penetration. This approach offers the potential for therapeutic synergy, where the combined effects on multiple targets produce greater overall benefit than would be predicted from the sum of individual activities, potentially allowing for lower effective doses and reduced side effect profile[29][30].
Pyrimidine as a Privileged Scaffold in Medicinal Chemistry
The pyrimidine ring represents one of the most ubiquitous and versatile heterocyclic scaffolds in medicinal chemistry, earning its designation as a “privileged structure” capable of interacting with a remarkable diversity of biological targets[31]. This nitrogen-containing six-membered heteroaromatic ring system forms the core of several essential biomolecules including the nucleic acid bases cytosine, thymine, and uracil, as well as numerous vitamins, cofactors, and nucleotides, reflecting its fundamental biological relevance and inherent compatibility with biological systems. The prevalence of pyrimidine in nature and its involvement in critical biochemical processes provide a compelling rationale for its therapeutic potential, as compounds incorporating this scaffold may benefit from evolved biological recognition and metabolic compatibility[32][33]. From a medicinal chemistry perspective, the pyrimidine ring offers exceptional opportunities for structural diversification through substitution at multiple positions, with the nitrogen atoms at positions 1 and 3 providing sites for hydrogen bonding interactions with biological targets while the carbon atoms at positions 2, 4, 5, and 6 accommodate various substituents that modulate physicochemical properties and target interactions [34][35]This structural versatility has been exploited in the development of approved drugs across numerous therapeutic areas, including antiviral agents such as zidovudine and acyclovir, anticancer drugs including imatinib and gefitinib, antihypertensive agents like minoxidil, and antipsychotics such as risperidone, demonstrating the scaffold’s remarkable adaptability and drug-like properties. In the specific context of Alzheimer’s disease research, pyrimidine-based compounds have demonstrated particular promise due to their ability to engage with multiple targets relevant to neurodegeneration[36][37]. The electron-rich aromatic system can participate in π-stacking interactions with aromatic residues in enzyme active sites, while the ring nitrogen atoms can coordinate metal ions implicated in amyloid aggregation and oxidative stress. Strategic introduction of appropriate substituents can confer antioxidant properties through radical scavenging mechanisms, enhance blood-brain barrier penetration by optimizing lipophilicity, and provide additional points of interaction with biological targets through hydrogen bonding, hydrophobic interactions, and electrostatic complementarity[38][39]. This combination of inherent biological relevance, structural versatility, synthetic accessibility, and demonstrated multi-target potential positions the pyrimidine scaffold as an ideal platform for the development of novel neuroprotective agents capable of addressing the multifactorial nature of Alzheimer’s disease pathology[40].
Pyrimidine Chemistry and Structural Basis for Neuroprotective Activity
Fundamental Structure and Physicochemical Properties of the Pyrimidine Ring
The pyrimidine ring system, with molecular formula C4H4N2, consists of a six-membered aromatic heterocycle containing two nitrogen atoms at the 1 and 3 positions, creating a symmetrical diazine structure with unique physicochemical characteristics that profoundly influence its biological interactions and drug-like properties[41][42]. The aromatic nature of the pyrimidine ring, arising from the conjugation of six π-electrons distributed over five atoms, provides structural rigidity and planarity that facilitates insertion into enzyme active sites and interaction with aromatic amino acid residues through π-stacking and π-cation interactions. The electronegative nitrogen atoms withdraw electron density from the ring through inductive effects while contributing electron density through resonance, creating a polarized electron distribution that influences both chemical reactivity and biological recognition. This electronic configuration results in a ring system that is electron-deficient relative to benzene, making it susceptible to nucleophilic attack while resistant to electrophilic substitution, directing synthetic strategies toward specific functionalization approaches. The presence of two nitrogen atoms also creates sites for hydrogen bonding, with the lone pair electrons on nitrogen serving as hydrogen bond acceptors while the ring C-H groups can participate as weak hydrogen bond donors, enabling multiple modes of interaction with biological macromolecules. The pyrimidine ring exhibits significant polarity, with calculated logP values typically lower than benzene or pyridine, contributing to aqueous solubility while still maintaining sufficient lipophilicity for membrane penetration when appropriately substituted. The pKa of the pyrimidine ring nitrogen atoms is influenced by substitution patterns and protonation states, with the N1 nitrogen being more basic than N3 due to resonance effects, and protonation occurring preferentially at lower pH to generate positively charged species that can interact with negatively charged amino acid residues[43][44]. This pH-dependent protonation behavior has implications for both target binding and pharmacokinetic properties, as the charged species may exhibit reduced membrane permeability while demonstrating enhanced aqueous solubility and altered target interactions. The pyrimidine ring demonstrates remarkable metabolic stability compared to many other heterocyclic systems, with the aromatic structure resisting facile oxidation or reduction under physiological conditions, although substituents attached to the ring may undergo various metabolic transformations. This combination of structural rigidity, electronic versatility, hydrogen bonding capacity, appropriate polarity, and metabolic stability makes the pyrimidine scaffold exceptionally well-suited for the development of central nervous system agents where balanced physicochemical properties are critical for achieving both target engagement and blood-brain barrier penetration[45][46].
Strategic Positions for Substitution and Structural Modifications
The pyrimidine nucleus offers five distinct positions for structural modification, each with unique reactivity patterns and differential impacts on biological activity, providing medicinal chemists with a versatile platform for fine-tuning pharmacological properties and target interactions. The C-2 position, located between the two nitrogen atoms, is particularly electron-deficient and susceptible to nucleophilic attack, making it a primary site for introducing diverse substituents through reactions with amines, alkoxides, and carbon nucleophiles. Substitutions at this position significantly influence both the electronic properties of the ring and its ability to engage in hydrogen bonding interactions, with the introduction of amino groups providing hydrogen bond donors and acceptors while alkyl or aryl substituents modulate lipophilicity and hydrophobic interactions[47]. The C-4 and C-6 positions, equivalent due to molecular symmetry in the unsubstituted parent compound, represent additional sites for functionalization with reactivity similar to but somewhat reduced compared to C-2, allowing for sequential introduction of different substituents to create asymmetrically substituted derivatives[48]. The C-5 position, uniquely lacking adjacent nitrogen atoms, exhibits distinct reactivity more closely resembling that of benzene, with somewhat greater susceptibility to electrophilic substitution and reduced sensitivity to nucleophilic attack, providing opportunities for introducing substituents that would be incompatible with the more electron-deficient positions. This differential reactivity enables sophisticated synthetic strategies where protecting groups and sequential functionalization can generate highly substituted pyrimidines with precisely controlled substitution patterns[49][50]. The strategic selection of substituents at these various positions allows medicinal chemists to systematically modulate physicochemical properties including lipophilicity, polarity, hydrogen bonding capacity, and molecular geometry, thereby optimizing blood-brain barrier penetration, metabolic stability, and target interactions. Substituents bearing hydrogen bond donors such as amino, hydroxyl, or amide groups at appropriate positions can establish critical interactions with enzyme active sites, while aromatic or heteroaromatic substituents can extend π-systems for enhanced stacking interactions with aromatic amino acid residues. The introduction of metal-chelating moieties, such as those present in Schiff bases, thiosemicarbazides, or hydroxamic acids, can confer the ability to bind redox-active metals implicated in oxidative stress and amyloid aggregation. Alkyl chains of varying lengths can modulate lipophilicity and provide additional hydrophobic interactions, while polar groups such as carboxylic acids or sulfonamides can influence solubility and pharmacokinetic profiles[51][52]. The three-dimensional arrangement of these substituents, influenced by the planar pyrimidine core and the conformational preferences of attached groups, determines the overall molecular shape and its complementarity to target binding sites, with the potential for creating molecules that can simultaneously engage multiple spatially distinct targets through appropriate spatial organization of pharmacophoric elements[53].
Pharmacophoric Features Contributing to Anti-AD Activity
The neuroprotective activity of pyrimidine-based compounds against Alzheimer’s disease pathology arises from the strategic incorporation of specific pharmacophoric features that enable interaction with the multiple biological targets implicated in neurodegeneration[54][55]. The pyrimidine core itself serves as more than a mere scaffold, actively contributing to target interactions through its aromatic system, which can engage in π-stacking with aromatic amino acid residues in enzyme active sites, particularly the catalytic anionic site of acetylcholinesterase where multiple aromatic residues create a hydrophobic environment for substrate binding[56][57]. The ring nitrogen atoms function as hydrogen bond acceptors, forming critical interactions with backbone amides or side chain hydroxyls in target proteins, while also participating in metal coordination when appropriately positioned relative to additional chelating groups. Electron-donating substituents, particularly amino groups at the C-2 or C-4 positions, enhance the electron density of the aromatic system and provide additional hydrogen bond donors, frequently improving cholinesterase inhibition by establishing interactions with the catalytic triad or peripheral anionic site. Electron-withdrawing groups can modulate the electronic properties of the ring and influence the acidity of adjacent protons, affecting both target interactions and physicochemical properties[58][59]. The incorporation of metal-chelating functionality represents a particularly valuable pharmacophoric feature for anti-Alzheimer’s activity, with Schiff base moieties formed by condensation of aminopyrimidines with aromatic aldehydes creating N,N or N,O donor sets capable of binding copper, iron, and zinc ions implicated in amyloid aggregation and oxidative stress. Thiosemicarbazide and hydrazone derivatives extend this chelating capacity while introducing additional hydrogen bonding possibilities and conformational flexibility that can enhance target interactions. Hydrophobic substituents, including aromatic rings and alkyl chains, contribute to membrane permeability and can occupy hydrophobic pockets in enzyme active sites, with the optimal balance of hydrophilic and hydrophobic features being critical for both potency and drug-like properties. The spatial arrangement of these pharmacophoric elements around the pyrimidine core determines the three-dimensional presentation of functional groups to biological targets, with the distance and orientation between critical features such as hydrogen bond donors, aromatic systems, and metal-chelating atoms being key determinants of multi-target activity. Molecular flexibility, introduced through appropriate linker regions, can allow the same molecule to adapt its conformation to engage with structurally distinct targets, while excessive flexibility may entropically disfavor binding and complicate structure-activity relationship interpretation. The strategic combination of these pharmacophoric features within a single pyrimidine-based molecule enables the creation of multi-target-directed ligands capable of simultaneously addressing cholinergic deficit, oxidative stress, metal dyshomeostasis, and protein aggregation, offering the potential for enhanced therapeutic efficacy through synergistic modulation of interconnected pathological pathways[60].
Influence of Substituents on Blood-Brain Barrier Permeability
The successful development of neurotherapeutic agents critically depends on achieving adequate blood-brain barrier penetration, as the central nervous system is protected by this highly selective endothelial barrier that restricts the passage of most circulating molecules into the brain parenchyma[61]. Pyrimidine-based compounds designed for Alzheimer’s therapy must navigate this formidable obstacle while maintaining the structural features necessary for target engagement, requiring careful optimization of physicochemical properties through strategic substitution. The blood-brain barrier permits passive diffusion primarily to small, lipophilic molecules with molecular weights below approximately 400-500 Daltons, and the pyrimidine core’s relatively low molecular weight provides an advantageous starting point for maintaining drug-like properties after necessary functionalization. Lipophilicity, commonly expressed as the logarithm of the octanol-water partition coefficient, represents a primary determinant of passive blood-brain barrier permeability, with optimal logP values for central nervous system penetration generally falling within the range of 1.5 to 3.0. Substituents that increase lipophilicity, such as halogen atoms, alkyl chains, and unsubstituted aromatic rings, generally enhance membrane permeability, while polar groups including carboxylic acids, sulfonates, and quaternary ammonium salts typically reduce penetration unless actively transported. However, excessive lipophilicity can lead to non-specific binding, reduced aqueous solubility, and increased metabolic clearance, necessitating careful balancing of hydrophobic and hydrophilic character. Hydrogen bonding capacity profoundly influences blood-brain barrier penetration, with each hydrogen bond donor or acceptor requiring energetic cost for desolvation during membrane partitioning, and compounds with fewer than eight hydrogen bond donors and acceptors combined generally exhibiting better permeability. The strategic placement of hydrogen bond donors and acceptors within the molecular structure can therefore significantly impact brain penetration, with the potential for intramolecular hydrogen bonding to mask polar functionality and effectively reduce the hydrogen bonding count experienced during membrane transit. Molecular flexibility and conformational preferences influence the presentation of polar functionality to the membrane environment, with rigid structures potentially adopting conformations that minimize polar surface area exposure. Total polar surface area, calculated from the sum of contributions of polar atoms, serves as a valuable predictor of blood-brain barrier permeability, with values below 60-70 Ų generally associated with good central nervous system penetration and values exceeding 90 Ų typically predicting limited brain access. The introduction of substituents that reduce polar surface area, such as halogenation of aromatic rings or conversion of polar functional groups to prodrug forms, can enhance permeability, while maintaining or introducing polar groups essential for target interactions presents a fundamental challenge in multi-target-directed ligand optimization[62-64]. The pyrimidine scaffold’s inherent polarity contributes to its polar surface area, but appropriate substitution can modulate this property within acceptable ranges while preserving the structural features required for neuroprotective activity. Active efflux by transporters such as P-glycoprotein represents an additional barrier that can limit brain penetration of otherwise permeable compounds, with certain structural features, including basic amines and specific aromatic substitution patterns, potentially serving as recognition elements for efflux transporters. Strategic modification to reduce transporter recognition while maintaining target interactions requires sophisticated understanding of structure-transport relationships and appropriate in vitro screening during lead optimization. The integration of blood-brain barrier permeability considerations from the earliest stages of compound design, rather than as an afterthought following the identification of potent target interactions, is essential for the successful development of clinically useful neurotherapeutic agents, ensuring that the molecules demonstrating promising in vitro activity can actually reach their intended sites of action within the central nervous system.[65]
Evolution from Single-Target to Multi-Target Design
The design paradigm for Alzheimer’s disease therapeutics has undergone a fundamental transformation over the past two decades, shifting from the classical “one molecule, one target” philosophy that dominated twentieth-century drug discovery toward a more sophisticated multi-target-directed approach that acknowledges the complex, multifactorial nature of neurodegenerative disorders. This evolution was driven by the repeated clinical failures of highly selective single-target agents that showed promising preclinical efficacy but failed to translate into meaningful therapeutic benefits in patients, highlighting the inadequacy of addressing only one component of a complex pathological network[66]. The single-target approach, rooted in Paul Ehrlich’s “magic bullet” concept, pursued highly selective inhibitors of acetylcholinesterase, secretases, or amyloid aggregation with the expectation that modulating a single rate-limiting step would interrupt the disease cascade. While this strategy yielded symptomatic treatments such as donepezil and rivastigmine, it failed to produce disease-modifying agents, suggesting that Alzheimer’s pathology involves redundant and interconnected pathways that can compensate for the inhibition of any single target. The recognition that amyloid-beta accumulation, tau hyperphosphorylation, oxidative stress, neuroinflammation, and metal dyshomeostasis operate as an interconnected network rather than a linear cascade provided the theoretical foundation for multi-target design[68][69]. In the context of pyrimidine-based compounds, this evolution manifests in the deliberate design of molecules capable of simultaneously engaging multiple targets through the strategic incorporation of diverse pharmacophoric elements around the central scaffold. Early pyrimidine derivatives were often optimized for a single activity, such as cholinesterase inhibition, with structural modifications focused on enhancing potency against that specific target. Contemporary design strategies instead seek to balance multiple activities, recognizing that moderate potency against several relevant targets may provide greater therapeutic benefit than exceptional potency against a single target. This paradigm shift has required corresponding changes in screening methodologies, with multi-target compounds evaluated across panels of assays rather than focused on a single endpoint, and structure-activity relationships interpreted in terms of multiple simultaneous optimizations. The pyrimidine scaffold has proven particularly amenable to this multi-target approach due to its synthetic versatility and the ability to attach diverse functional groups that engage different targets through distinct binding modes, enabling the creation of molecules that can, for example, simultaneously inhibit cholinesterase enzymes, chelate redox-active metals, and scavenge free radicals through complementary structural elements[70].
Hybrid Molecule Design: Combining Pyrimidine with Other Pharmacophores
Hybrid molecule design represents a particularly fruitful strategy for creating multi-target pyrimidine-based compounds, involving the covalent linkage of the pyrimidine core with other pharmacophoric fragments known to interact with targets relevant to Alzheimer’s disease pathology[71][72]. This approach draws inspiration from natural product hybrids and has been systematically applied to combine the favorable properties of pyrimidine with those of other privileged structures, creating molecules that inherit and potentially synergize the activities of both components. The design process typically begins with the identification of two or more pharmacophores with complementary target profiles, such as a pyrimidine-based cholinesterase inhibitor and an antioxidant fragment derived from natural polyphenols, followed by their connection through appropriate linker regions that maintain the essential binding features of each component while allowing sufficient conformational flexibility for simultaneous target engagement. The choice of linker is critical, as it must position the pharmacophores at appropriate distances and orientations relative to their respective binding sites while avoiding the introduction of metabolic liabilities or excessive molecular weight that would compromise drug-like properties[73]. Common linkers include alkyl chains of varying lengths, which provide flexibility and hydrophobic character, amide or ester linkages that introduce hydrogen bonding capacity, and triazole rings formed through click chemistry that combine structural rigidity with synthetic accessibility. The pyrimidine nucleus can serve as the central scaffold to which multiple pharmacophoric fragments are attached, or it can itself function as one of the pharmacophoric elements in a hybrid structure where another core carries the pyrimidine as a substituent. Examples of successful hybrid designs include pyrimidine-chalcone conjugates combining the enzyme inhibitory potential of pyrimidine with the antioxidant and anti-inflammatory properties of chalcones, pyrimidine-coumarin hybrids leveraging the metal-chelating and antioxidant activities of coumarins alongside pyrimidine’s enzyme inhibition capacity, and pyrimidine-tacrine hybrids that combine the potent cholinesterase inhibition of tacrine with the favorable safety profile and additional target interactions of pyrimidine derivatives[74]. The hybridization approach also enables the incorporation of fragments specifically designed to enhance blood-brain barrier penetration, such as lipophilic amino acid esters or glucose derivatives that may exploit nutrient transporters for active uptake into the central nervous system. Structure-activity relationship studies of hybrid molecules have revealed that the optimal balance of activities often requires systematic variation of linker length and composition, as well as the precise points of attachment on each pharmacophore, with even minor structural changes significantly impacting the multi-target profile. The synthetic accessibility of hybrid molecules through modular assembly strategies, often employing parallel synthesis or combinatorial approaches, facilitates the rapid exploration of chemical space around successful hybrid designs and accelerates the identification of optimized leads with balanced multi-target activities and favorable drug-like properties[75].
Metal Complexation Strategies: Pyrimidine-Based Schiff Base Complexes
The complexation of pyrimidine-based ligands with transition metal ions represents a sophisticated design strategy that exploits the coordination chemistry of the pyrimidine scaffold to create compounds with enhanced and mechanistically distinct neuroprotective properties[76][77]. This approach is grounded in the recognized role of metal dyshomeostasis in Alzheimer’s pathology, where elevated concentrations of copper, iron, and zinc within senile plaques contribute to amyloid-beta aggregation, generate reactive oxygen species through Fenton chemistry, and exacerbate oxidative stress and neuroinflammation[78][79]. Pyrimidine-based Schiff bases, formed by condensation of aminopyrimidines with aromatic or heteroaromatic aldehydes, create versatile ligand systems with nitrogen and oxygen donor atoms ideally positioned for metal coordination, enabling the formation of stable complexes with copper(II), iron(III), zinc(II), and other biologically relevant metal ions. The design of these complexes must consider multiple factors including the geometry preferences of the metal center, the denticity and spatial arrangement of donor atoms, the overall charge and lipophilicity of the resulting complex, and the kinetic and thermodynamic stability under physiological conditions. Mononuclear complexes featuring a single metal center coordinated by one or more pyrimidine-based ligands represent the most common design, with the metal oxidation state and coordination geometry influencing both the stability of the complex and its biological interactions. Bimetallic and polynuclear complexes offer additional structural diversity and potential for enhanced metal-buffering capacity, though their larger molecular size may compromise blood-brain barrier penetration[80]. The metal center itself can contribute to neuroprotective activity through multiple mechanisms: direct sequestration of excess metal ions that would otherwise participate in pathological processes, catalytic antioxidant activity mimicking superoxide dismutase or catalase, modulation of amyloid-beta aggregation through metal coordination, and generation of biologically active species through redox cycling under controlled conditions. Platinum(II) and ruthenium(II) complexes of pyrimidine-based ligands have attracted particular attention due to the established anticancer activity of these metal centers and emerging evidence for their neuroprotective potential, with studies demonstrating that complexation can enhance cholinesterase inhibition, improve antioxidant capacity, and confer anti-amyloidogenic properties compared to the free ligands[81]. The design of metal complexes also offers opportunities for targeting specific subcellular compartments, with lipophilic complexes potentially accumulating in mitochondria where they can modulate oxidative phosphorylation and reduce reactive oxygen species generation at their source. Characterization of these complexes requires sophisticated analytical techniques including UV-Vis and circular dichroism spectroscopy to elucidate coordination geometry, electron paramagnetic resonance for paramagnetic metal centers, cyclic voltammetry to assess redox properties, and mass spectrometry to confirm stoichiometry and identify potential solution-state speciation. The stability of metal complexes under physiological conditions, particularly in the presence of competing biological ligands such as albumin, glutathione, and histidine-rich proteins, represents a critical consideration that must be addressed through appropriate design and rigorous in vitro evaluation to ensure that the intact complex, rather than dissociated components, mediates the observed biological effects[82][83].
Pharmacophore Modeling and Virtual Screening
Pharmacophore modeling and virtual screening have emerged as indispensable computational tools in the rational design of pyrimidine-based multi-target-directed ligands for Alzheimer’s therapy, enabling the efficient exploration of vast chemical space and the identification of novel scaffolds with optimal combinations of target interactions[84]. A pharmacophore model represents the three-dimensional arrangement of steric and electronic features essential for biological activity, typically including hydrogen bond donors and acceptors, hydrophobic regions, aromatic rings, positively or negatively ionizable groups, and metal-binding motifs, all positioned at specific distances and angles relative to each other. These models can be constructed through structure-based approaches when three-dimensional structures of target proteins are available, extracting interaction features from protein-ligand complexes, or through ligand-based methods that align and compare sets of known active compounds to identify common pharmacophoric patterns. For Alzheimer’s disease applications, pharmacophore models have been developed for multiple relevant targets including acetylcholinesterase, butyrylcholinesterase, beta-secretase, monoamine oxidase, and various metal-binding sites, enabling the design of compounds that simultaneously satisfy the feature requirements of several targets[85]. The pyrimidine scaffold’s well-defined three-dimensional structure and multiple substitution sites make it particularly amenable to pharmacophore-based design, as the core can serve as a rigid template for orienting pharmacophoric features in specific spatial arrangements required for multi-target engagement. Virtual screening employs these pharmacophore models as queries to search large compound libraries, either commercially available or virtual, rapidly identifying molecules that contain the necessary features in the appropriate three-dimensional arrangement[86]. This computational filtering can reduce millions of compounds to manageable numbers of high-potential candidates for experimental evaluation, dramatically accelerating the discovery process and reducing the resources required for hit identification. Structure-based virtual screening extends this approach by docking compounds into the three-dimensional structures of target proteins, calculating binding energies and interaction patterns to prioritize molecules with favorable predicted binding modes. For multi-target applications, compounds can be screened sequentially against multiple targets, with only those showing acceptable predicted interactions with all desired targets advancing for experimental evaluation. Machine learning and artificial intelligence approaches are increasingly integrated with pharmacophore modeling, using trained models to predict activities against multiple targets based on molecular descriptors and structural features, enabling the rapid assessment of vast virtual libraries and the generation of novel structures with optimized multi-target profiles[87][88]. The integration of pharmacophore modeling with other computational tools, including molecular dynamics simulations to assess binding stability, ADMET prediction to evaluate drug-like properties, and synthetic accessibility scoring to ensure practical realizability, creates a comprehensive computational framework that guides experimental efforts toward the most promising pyrimidine-based candidates. Successful applications of this approach in Alzheimer’s drug discovery have identified novel pyrimidine derivatives with balanced cholinesterase inhibition and antioxidant activity, compounds combining BACE-1 inhibition with metal chelation, and molecules that simultaneously target amyloid aggregation and neuroinflammation, demonstrating the power of computational design in addressing the multifactorial nature of neurodegenerative disease[88][89].
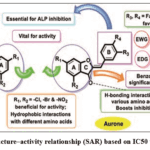 |
Figure 2: Combined structure–activity relationship (SAR) based on IC50 values and docking study Click here to View Figure |
Synthesis and Characterization of Pyrimidine Derivatives
The synthesis of functionalized pyrimidine derivatives for neuroprotective evaluation encompasses a diverse array of established and innovative methodologies that enable the introduction of substituents at all positions of the heterocyclic core with precise control over regiochemistry and stereochemistry. Classical approaches to pyrimidine synthesis include the Biginelli reaction, which provides convergent access to 3,4-dihydropyrimidin-2(1H)-ones through one-pot condensation of β-ketoesters, aldehydes, and urea or thiourea derivatives, offering a practical route to diverse pyrimidine libraries with variations at multiple positions. The Pinner synthesis, involving the condensation of amidines with β-dicarbonyl compounds or their equivalents, provides another versatile approach that has been extensively employed for the preparation of substituted pyrimidines with controlled substitution patterns[90]. Modern variations of these classical methods employ microwave irradiation, ultrasound assistance, and flow chemistry techniques to enhance reaction rates, improve yields, and reduce environmental impact, enabling rapid access to diverse compound libraries for structure-activity relationship studies. Transition metal-catalyzed cross-coupling reactions, particularly Suzuki-Miyaura, Sonogashira, and Buchwald-Hartwig couplings, have revolutionized pyrimidine functionalization by enabling the direct introduction of aryl, alkynyl, and amino substituents at halogenated positions under mild conditions with exceptional functional group tolerance. The selective halogenation of pyrimidine precursors at specific positions, achieved through directed ortho-metalation or electrophilic substitution under carefully controlled conditions, provides versatile intermediates for subsequent cross-coupling diversification. Nucleophilic aromatic substitution exploits the electron-deficient nature of the pyrimidine ring, particularly at positions activated by adjacent nitrogen atoms, allowing direct displacement of leaving groups such as halides or sulfonates by amines, alkoxides, and carbon nucleophiles under relatively mild conditions. Solid-phase synthesis and combinatorial chemistry approaches have been adapted for pyrimidine diversification, enabling the parallel synthesis of large compound libraries through split-and-pool strategies or multi-step sequences on polymeric supports with automated purification and analysis. The choice of synthetic route depends on multiple factors including the desired substitution pattern, the availability of starting materials, the scale required for biological evaluation, and the need for enantioselective synthesis when chiral centers are introduced. Protection-deprotection strategies are frequently necessary when multiple functional groups require sequential manipulation, with silyl ethers, acetals, and tert-butoxycarbonyl groups commonly employed for temporary protection of sensitive functionality. The optimization of synthetic routes for lead compounds identified through screening programs typically focuses on scalability, cost-effectiveness, and environmental sustainability, with telescoped sequences that minimize isolation of intermediates and reduce solvent consumption being particularly valuable for kilogram-scale preparations required for advanced preclinical studies. Recent advances in photoredox catalysis and electrochemical synthesis have opened new opportunities for pyrimidine functionalization under mild, environmentally friendly conditions, enabling transformations that were previously difficult or impossible to achieve, and expanding the accessible chemical space for neuroprotective agent discovery[91].
Synthesis of Pyrimidine-Based Schiff Bases and Their Metal Complexes
Condensation Reactions with Aromatic Aldehydes
The synthesis of pyrimidine-based Schiff bases proceeds through the classical condensation reaction between primary amine-functionalized pyrimidines and aromatic or heteroaromatic aldehydes, representing one of the most straightforward and versatile approaches for generating nitrogen-containing ligands with excellent metal-coordinating capabilities. The reaction mechanism involves nucleophilic attack of the pyrimidine amine on the electrophilic aldehyde carbonyl carbon, forming a carbinolamine intermediate that subsequently undergoes dehydration to yield the characteristic azomethine linkage with elimination of water, establishing a carbon-nitrogen double bond that serves as the primary metal coordination site alongside additional donor atoms from the aldehyde component[92]. The choice of aminopyrimidine precursor significantly influences the reactivity and yield of the condensation reaction, with 2-aminopyrimidine, 4-aminopyrimidine, 5-aminopyrimidine, and 2,4-diaminopyrimidine each offering distinct substitution patterns that affect both the electronic properties of the resulting Schiff base and the spatial arrangement of donor atoms available for metal coordination. Aromatic aldehydes bearing various substituents, including salicylaldehyde with its ortho-hydroxyl group providing an additional oxygen donor atom for bidentate or tridentate coordination, pyridinecarboxaldehydes introducing heterocyclic nitrogen donors, and hydroxy-naphthaldehydes extending the π-system for enhanced aromatic interactions, enable systematic variation of the ligand environment and fine-tuning of metal complex properties through electronic and steric modulation. The condensation reaction is typically conducted in refluxing alcoholic solvents such as methanol, ethanol, or isopropanol, often with the addition of catalytic amounts of acetic acid or p-toluenesulfonic acid to accelerate imine formation by protonating the carbonyl oxygen and enhancing electrophilicity, while azeotropic removal of water using molecular sieves or Dean-Stark apparatus drives the equilibrium toward product formation[93]. Reaction progress is conveniently monitored by thin-layer chromatography, with the appearance of new, less polar spots corresponding to Schiff base formation, and by infrared spectroscopy where the disappearance of the aldehyde carbonyl stretch near 1700 cm⁻¹ and the amine N-H stretches accompanies the appearance of the characteristic imine C=N absorption around 1620-1640 cm⁻¹. Microwave-assisted synthesis has been successfully applied to pyrimidine Schiff base formation, reducing reaction times from hours to minutes while often improving yields and product purity through rapid, uniform heating and enhanced reaction kinetics under controlled pressure and temperature conditions[94]. The resulting Schiff bases typically precipitate from the reaction mixture upon cooling or after solvent concentration, allowing isolation by simple filtration and purification by recrystallization from appropriate solvents or by column chromatography when necessary to remove unreacted starting materials or side products. Structural characterization of the synthesized Schiff bases employs multiple complementary techniques including proton and carbon-13 nuclear magnetic resonance spectroscopy, where the characteristic imine proton appears as a singlet in the 8-9 ppm region and the imine carbon resonates between 150-165 ppm, infrared spectroscopy confirming imine formation through the C=N stretch, and high-resolution mass spectrometry providing unambiguous confirmation of molecular composition[95][96]. The geometric configuration of the imine bond typically adopts the thermodynamically favored trans conformation, though cis isomers may be observed in constrained systems or upon metal complexation, and this conformational preference influences both the ligand’s metal-binding properties and the three-dimensional structure of resulting complexes. The synthetic accessibility of pyrimidine Schiff bases, combined with the extensive variability achievable through different amine and aldehyde combinations, has made this class of compounds particularly valuable for exploring structure-activity relationships in neuroprotective agent development and for generating libraries of ligands with systematically varied electronic and steric properties for metal complexation studies[97].
Complexation with Platinum(II) and Ruthenium(II) Ions
The complexation of pyrimidine-based Schiff base ligands with platinum(II) and ruthenium(II) ions represents a sophisticated strategy for generating metal-based neuroprotective agents with enhanced and mechanistically distinct biological activities compared to their parent organic frameworks[98]. The coordination chemistry of these precious metal ions with Schiff base ligands exploits the electron-rich nature of the azomethine nitrogen and additional donor atoms from the aldehyde component, typically oxygen from phenolic hydroxyls or nitrogen from pyridine rings, creating stable chelate rings that confer thermodynamic stability and kinetic inertness under physiological conditions. Platinum(II) complexes are typically prepared by reacting the Schiff base ligand with potassium tetrachloroplatinate(II) or dichlorobis(dimethylsulfoxide)platinum(II) in appropriate solvents, with the reaction conditions carefully controlled to favor the formation of square planar complexes where the ligand occupies two coordination sites through bidentate binding, with the remaining sites occupied by chloride or other ancillary ligands that influence solubility and biological activity. The choice of platinum precursor and reaction stoichiometry determines whether neutral complexes of the type [Pt(L)Cl₂] or cationic complexes such as [Pt(L)(solvent)₂]²⁺ are formed, with each exhibiting distinct solubility, cellular uptake, and DNA-binding properties that impact their neuroprotective potential.[99] Ruthenium(II) complexes offer greater structural diversity due to the metal’s ability to adopt octahedral coordination geometry and accommodate a wider range of ligand types, with synthetic approaches typically employing ruthenium precursors such as dichlorotetrakis(dimethylsulfoxide)ruthenium(II) or dichloro(p-cymene)ruthenium(II) dimer that allow systematic variation of the coordination sphere. The reaction of pyrimidine Schiff bases with ruthenium precursors proceeds through stepwise ligand substitution, with the final product geometry determined by the steric demands of the ligand, the nature of ancillary ligands, and reaction conditions including temperature, solvent, and the presence of coordinating or non-coordinating counterions. Half-sandwich ruthenium(II) arene complexes, where the Schiff base ligand occupies two or three coordination sites and an arene ring such as p-cymene or benzene caps one face of the octahedron, have attracted particular attention due to their favorable balance of stability and reactivity, tunable lipophilicity through arene modification, and potential for synergistic interactions between the metal center, arene ligand, and Schiff base framework. Characterization of the resulting metal complexes requires sophisticated analytical techniques to confirm coordination geometry, oxidation state, and purity, with single-crystal X-ray diffraction providing definitive structural information including bond lengths and angles, coordination geometry, and the spatial arrangement of ligands around the metal center. Spectroscopic characterization employs infrared spectroscopy to monitor shifts in the characteristic imine C=N stretch upon coordination, typically moving to lower wavenumbers by 10-30 cm⁻¹ due to reduced bond order upon nitrogen donation to the metal, and nuclear magnetic resonance spectroscopy where paramagnetic metals preclude analysis but diamagnetic platinum(II) and low-spin ruthenium(II)[95] complexes yield well-resolved spectra with characteristic coordination-induced shifts. UV-Visible spectroscopy reveals the d-d transitions and metal-to-ligand charge transfer bands characteristic of each metal complex, providing information about electronic structure and serving as a fingerprint for compound identity and purity. Mass spectrometry, particularly electrospray ionization and matrix-assisted laser desorption ionization techniques, confirms complex stoichiometry and can provide insight into solution speciation and the presence of aggregates or higher nuclearity species. Electrochemical characterization through cyclic voltammetry reveals the redox behavior of the metal center and ligand framework, with the oxidation and reduction potentials providing insight into the electronic properties that may influence antioxidant activity and the ability to participate in biological redox cycling[96]. The stability of these metal complexes under physiological conditions, particularly in the presence of competing biological ligands such as glutathione, albumin, and histidine, must be rigorously evaluated through solution studies using UV-Vis spectroscopy or high-performance liquid chromatography to monitor complex integrity over time and under conditions that mimic the biological milieu. Biological evaluation of platinum(II) and ruthenium(II) pyrimidine Schiff base complexes has revealed enhanced cholinesterase inhibition compared to free ligands, improved antioxidant capacity through both direct radical scavenging and catalytic antioxidant mechanisms, and promising anti-amyloidogenic properties through metal-mediated modulation of peptide aggregation, supporting continued investigation of these metal-based agents for Alzheimer’s therapy[97].
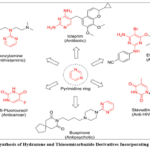 |
Figure 3; Synthesis of Hydrazone and Thiosemicarbazide Derivatives Incorporating Pyrimidine Click here to View Figure |
The synthesis of hydrazone and thiosemicarbazide derivatives incorporating the pyrimidine scaffold provides access to compounds with exceptional metal-chelating capacity, diverse biological activities, and structural features that enable multiple modes of interaction with Alzheimer’s disease-related targets. Hydrazone derivatives are typically prepared through condensation reactions between pyrimidine carboxaldehydes or ketones and variously substituted hydrazines or hydrazides, or alternatively through the reaction of hydrazine-functionalized pyrimidines with carbonyl compounds, providing symmetrical access to this important class of compounds. The reaction proceeds through nucleophilic addition of the hydrazine nitrogen to the carbonyl carbon, forming a tetrahedral intermediate that undergoes dehydration to yield the stable hydrazone linkage, with the reaction typically conducted in refluxing ethanol or methanol, often with catalytic acetic acid to accelerate imine formation and azeotropic removal of water to drive the equilibrium toward product formation. Aromatic hydrazides bearing additional functional groups, including isonicotinic acid hydrazide with its pyridine ring providing an additional metal coordination site, benzoic acid hydrazides with various ring substituents modulating electronic properties, and carbohydrate-derived hydrazides introducing hydrophilic character, enable systematic variation of the hydrazone component and fine-tuning of molecular properties. Thiosemicarbazide derivatives incorporate the N-N-C(S)-N functional group, combining the chelating capacity of thiourea with the additional donor atoms and conformational flexibility of the hydrazine linkage, and are typically synthesized through the reaction of pyrimidine isothiocyanates with hydrazines, or alternatively through the condensation of pyrimidine thiosemicarbazides with carbonyl compounds to generate thiosemicarbazones with extended conjugation. The synthesis of pyrimidine thiosemicarbazides often proceeds through the initial formation of pyrimidine isothiocyanates from the corresponding amines using thiophosgene or its safer alternatives such as thiocarbonyldiimidazole or phenyl chlorothionoformate, followed by reaction with hydrazine or substituted hydrazines under mild conditions to yield the desired products. The resulting hydrazone and thiosemicarbazide derivatives exhibit characteristic spectroscopic features including infrared absorptions for NH stretches in the 3100-3400 cm⁻¹ region, carbonyl stretches for hydrazides near 1650-1700 cm⁻¹, and thiocarbonyl stretches for thiosemicarbazides around 1200-1350 cm⁻¹. Nuclear magnetic resonance spectroscopy reveals the hydrazone NH proton as a distinctive broad singlet in the 8-12 ppm region, often showing exchange broadening and temperature-dependent chemical shifts, while the azomethine proton in hydrazones appears as a singlet between 7.5-8.5 ppm with chemical shifts sensitive to conjugation and substitution patterns. Carbon-13 NMR shows the hydrazone imine carbon in the 140-160 ppm range, the hydrazide carbonyl near 160-175 ppm, and the thiosemicarbazide thiocarbonyl around 175-185 ppm, providing diagnostic signals for structure confirmation. The metal-chelating properties of these derivatives arise from the combination of donor atoms including the pyrimidine nitrogens, the hydrazone imine nitrogen, the hydrazide carbonyl oxygen or thiosemicarbazide sulfur, and additional donor atoms from substituents, creating versatile ligand systems capable of forming stable complexes with copper, iron, and zinc ions implicated in Alzheimer’s pathology. The conformational flexibility imparted by the hydrazone linkage allows these molecules to adapt their geometry to accommodate different metal coordination preferences and to present chelating groups optimally for metal binding, while also enabling interactions with enzyme active sites through induced fit mechanisms. The synthetic accessibility of pyrimidine hydrazone and thiosemicarbazide derivatives through modular assembly from commercially available precursors, combined with their demonstrated potential as multi-target neuroprotective agents with cholinesterase inhibitory, antioxidant, and anti-amyloidogenic activities, supports continued interest in this compound class for Alzheimer’s drug discovery.
In Vitro Neuroprotective Evaluation: Methodologies and Key Findings
Cholinesterase Inhibition Assays
The evaluation of cholinesterase inhibitory activity represents a cornerstone of in vitro neuroprotective assessment for pyrimidine-based compounds targeting Alzheimer’s disease, reflecting the well-established role of cholinergic deficit in disease symptomatology and the clinical validation of cholinesterase inhibition as a therapeutic strategy. The cholinergic hypothesis of Alzheimer’s disease posits that degeneration of basal forebrain cholinergic neurons and consequent depletion of acetylcholine in cortical and hippocampal regions contributes significantly to cognitive decline, making the restoration of cholinergic neurotransmission through inhibition of acetylcholine-hydrolyzing enzymes a rational therapeutic approach. Two distinct cholinesterase enzymes exist in humans: acetylcholinesterase, which is primarily responsible for terminating neurotransmission at cholinergic synapses through rapid hydrolysis of acetylcholine, and butyrylcholinesterase, a broader specificity enzyme that also hydrolyzes acetylcholine and may play compensatory roles when acetylcholinesterase activity is compromised[101][102]. The standard methodology for assessing cholinesterase inhibition employs the Ellman colorimetric assay, a spectrophotometric method that has become the gold standard due to its simplicity, reproducibility, and suitability for high-throughput screening applications. This assay utilizes acetylthiocholine or butyrylthiocholine as substrates that mimic the natural neurotransmitter, with enzymatic hydrolysis generating thiocholine that reacts with 5,5′-dithiobis-(2-nitrobenzoic acid) to produce the yellow-colored 5-thio-2-nitrobenzoate anion, whose absorbance is monitored at 412 nm over time. The rate of absorbance increase is directly proportional to enzyme activity, and the reduction in this rate in the presence of test compounds provides a quantitative measure of inhibition, typically expressed as percentage inhibition at specific concentrations or as IC₅₀ values representing the concentration required to achieve 50% enzyme inhibition. The assay is conducted in multi-well plate formats, enabling simultaneous evaluation of multiple compounds across concentration ranges, with appropriate controls including blank wells without enzyme to correct for non-enzymatic substrate hydrolysis and positive controls using reference inhibitors such as donepezil, rivastigmine, or galantamine for validation. Enzyme sources include electric eel acetylcholinesterase and equine serum butyrylcholinesterase for initial screening, though confirmation using human recombinant enzymes is essential for compounds intended for clinical development due to potential species differences in enzyme structure and inhibitor sensitivity[105]. Kinetic studies employing varying substrate concentrations in the presence of fixed inhibitor concentrations enable determination of inhibition mechanisms, distinguishing between competitive inhibition where the compound binds to the active site, non-competitive inhibition where binding occurs at peripheral sites, and mixed-type inhibition combining both mechanisms. The interpretation of cholinesterase inhibition data must consider factors including compound solubility in assay buffers, potential interference with the Ellman reagent through thiol reactivity or colorimetric interference, and the possibility of time-dependent inhibition requiring pre-incubation with enzyme before substrate addition. Structure-activity relationships derived from cholinesterase inhibition assays have guided the optimization of pyrimidine-based compounds, revealing that electron-donating substituents, particularly amino groups at the C-2 and C-4 positions, generally enhance acetylcholinesterase inhibition, while more lipophilic substituents favor butyrylcholinesterase interaction. The therapeutic relevance of cholinesterase inhibition extends beyond symptomatic cognitive enhancement, with emerging evidence suggesting that acetylcholinesterase may directly modulate amyloid-beta aggregation through peripheral anionic site interactions, and that butyrylcholinesterase inhibition becomes increasingly important as disease progresses and acetylcholinesterase levels decline, supporting the development of compounds with balanced or selective inhibition profiles optimized for different disease stages[106].
Acetylcholinesterase (AChE) Inhibitory Activity
The evaluation of acetylcholinesterase inhibitory activity for pyrimidine-based compounds reveals the critical structural features that govern interaction with this clinically validated target, providing fundamental insights that guide medicinal chemistry optimization and lead compound selection[107]. Acetylcholinesterase possesses a complex active site architecture comprising the catalytic anionic site at the base of a deep gorge, where the catalytic triad of serine, histidine, and glutamate performs ester hydrolysis, and the peripheral anionic site near the gorge entrance, which serves as an allosteric modulation site and has been implicated in amyloid-beta interactions. Pyrimidine derivatives engage with these sites through multiple binding modes, with the planar heterocyclic core participating in π-stacking interactions with the numerous aromatic residues lining the active site gorge, particularly tryptophan 84 at the catalytic anionic site and tryptophan 279 at the peripheral site. The inhibitory potency of pyrimidine-based compounds against acetylcholinesterase is typically expressed as IC₅₀ values determined through Ellman assays, with lead compounds demonstrating values in the nanomolar to low micromolar range comparable to or exceeding clinically used drugs. Structure-activity relationship studies across diverse pyrimidine derivative classes reveal that 2-aminopyrimidine substructures consistently enhance acetylcholinesterase inhibition, with the amino group providing hydrogen bond donation to active site residues and increasing electron density for enhanced π-stacking interactions. Substitutions at the C-4 and C-6 positions with aromatic or heteroaromatic rings extend molecular reach along the active site gorge, enabling simultaneous interaction with both catalytic and peripheral sites, a binding mode associated with particularly potent inhibition and potential amyloid-beta anti-aggregation properties. The introduction of basic nitrogen centers, such as those in piperazine or aminoalkyl side chains, can enhance binding through cation-π interactions with aromatic residues and ionic interactions with the negatively charged gorge entrance.[107]. Pyrimidinylthiourea derivatives have demonstrated exceptional acetylcholinesterase inhibitory activity, with certain compounds achieving IC₅₀ values in the nanomolar range through combined interactions of the pyrimidine core with the catalytic site, the thiourea sulfur with the peripheral site, and hydrogen bonding networks involving the thiourea NH groups. Phthalocyanine derivatives incorporating pyrimidine substituents show concentration-dependent acetylcholinesterase inhibition, with the large macrocyclic platform enabling simultaneous engagement of multiple enzyme molecules or occupation of extended binding surfaces inaccessible to smaller compounds. Hydrazone and thiosemicarbazide derivatives exhibit acetylcholinesterase inhibition that correlates with the electron-withdrawing or electron-donating character of substituents on the aldehyde component, with methoxy and hydroxy substitutions generally enhancing activity through additional hydrogen bonding interactions. The mechanism of acetylcholinesterase inhibition for pyrimidine compounds is typically competitive or mixed-type, reflecting binding at the catalytic site with additional interactions at peripheral regions, and can be elucidated through Lineweaver-Burk plot analysis of kinetic data. Molecular docking studies complement experimental inhibition data by predicting binding orientations and interactions, revealing that active compounds typically position the pyrimidine core near the catalytic triad while directing substituents along the gorge toward the peripheral site or into hydrophobic pockets. The therapeutic relevance of potent acetylcholinesterase inhibition must be balanced with selectivity considerations, as excessive inhibition at peripheral sites can lead to cholinergic side effects including gastrointestinal distress and bradycardia, supporting the pursuit of compounds with optimized inhibition profiles rather than maximal potency[108].
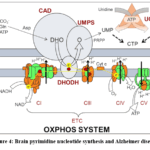 |
Figure 4: Brain pyrimidine nucleotide synthesis and Alzheimer disease Click here to View table |
Butyrylcholinesterase (BChE) Inhibitory Activity
The evaluation of butyrylcholinesterase inhibitory activity has gained increasing importance in Alzheimer’s drug discovery, reflecting recognition of this enzyme’s distinct and complementary role in cholinergic neurotransmission and its potential as a therapeutic target, particularly in advanced disease stages[1058]. Butyrylcholinesterase, also known as pseudocholinesterase or plasma cholinesterase, is synthesized in the liver and widely distributed in the body, including within the central nervous system where it is localized to glial cells and specific neuronal populations. Unlike acetylcholinesterase, which shows strict substrate specificity for acetylcholine, butyrylcholinesterase hydrolyzes a broader range of choline esters and exhibits different kinetic properties, including resistance to substrate inhibition and the ability to function at higher substrate concentrations[109]. In the healthy brain, acetylcholinesterase predominates and carries the majority of acetylcholine hydrolysis, but as Alzheimer’s disease progresses, acetylcholinesterase levels in specific brain regions decline by up to 45%, while butyrylcholinesterase activity progressively increases, suggesting a compensatory role that makes butyrylcholinesterase inhibition increasingly relevant for maintaining cholinergic function in moderate to severe disease. The structural basis for butyrylcholinesterase inhibition differs from acetylcholinesterase due to amino acid substitutions in the active site gorge, particularly the replacement of aromatic residues with smaller aliphatic amino acids that create a larger, more accommodating binding pocket capable of accepting bulkier inhibitors. Pyrimidine-based compounds demonstrate variable butyrylcholinesterase inhibitory activity depending on their size, shape, and substitution patterns, with generally more lipophilic and bulky derivatives showing enhanced potency against this enzyme. Structure-activity relationship studies reveal that butyrylcholinesterase inhibition by pyrimidine derivatives often correlates with different structural features than those governing acetylcholinesterase activity, with alkyl substitutions and extended aromatic systems favoring butyrylcholinesterase interaction while polar hydrogen-bonding groups may be less critical. Pyrimidinylthiourea derivatives have shown promising butyrylcholinesterase inhibition with selectivity profiles that can be tuned through modification of the thiourea substituent, with larger aromatic groups generally favoring butyrylcholinesterase over acetylcholinesterase. Phthalocyanine derivatives exhibit butyrylcholinesterase inhibition that may involve different binding modes than their acetylcholinesterase interactions, with the large macrocyclic platform potentially occupying the expanded butyrylcholinesterase gorge in orientations distinct from those adopted in the narrower acetylcholinesterase active site. Hydrazone derivatives demonstrate butyrylcholinesterase inhibition that can be optimized independently from acetylcholinesterase activity through appropriate substitution, with certain compounds achieving balanced dual inhibition while others show pronounced selectivity for one enzyme over the other. The therapeutic implications of butyrylcholinesterase-selective inhibition versus balanced dual inhibition remain under investigation, with evidence supporting both approaches depending on disease stage and individual patient characteristics. Selective butyrylcholinesterase inhibitors may offer advantages in early disease by preserving acetylcholinesterase activity for cognitive functions while providing additional cholinergic support, while dual inhibitors may be more appropriate for advanced disease where both enzymes contribute significantly to acetylcholine hydrolysis. The Ellman assay for butyrylcholinesterase inhibition employs butyrylthiocholine as substrate with equine serum or human recombinant enzyme, following the same spectrophotometric principles as acetylcholinesterase evaluation but requiring distinct positive controls such as ethopropazine or selective butyrylcholinesterase inhibitors for validation. Kinetic analysis of butyrylcholinesterase inhibition mechanisms for pyrimidine compounds typically reveals competitive or mixed-type inhibition, with the larger active site accommodating diverse binding modes that may involve interactions with both catalytic and peripheral regions. The correlation between in vitro butyrylcholinesterase inhibition and in vivo cognitive enhancement has been established through studies with selective inhibitors, supporting the therapeutic relevance of this target and the continued optimization of pyrimidine-based compounds for butyrylcholinesterase activity alongside other neuroprotective properties.
Selectivity Index and Structure-Activity Correlations
The selectivity index, calculated as the ratio of butyrylcholinesterase IC₅₀ to acetylcholinesterase IC₅₀ or vice versa, provides a quantitative measure of an inhibitor’s preference for one cholinesterase enzyme over the other and represents a critical parameter in the optimization of pyrimidine-based compounds for Alzheimer’s therapy. Compounds with selectivity indices greater than one indicate preference for acetylcholinesterase inhibition when calculated as BChE IC₅₀/AChE IC₅₀, while values less than one indicate butyrylcholinesterase preference, with the magnitude reflecting the degree of selectivity. The therapeutic implications of selectivity profile remain subject to ongoing investigation, with evidence supporting different optimal profiles depending on disease stage, patient genetics, and the specific combination of additional activities incorporated into multi-target compounds. Structure-activity relationship analysis across diverse pyrimidine derivative classes reveals systematic correlations between molecular features and cholinesterase selectivity that guide rational design toward desired profiles. Electron-donating substituents, particularly amino groups at the C-2 and C-4 positions, consistently favor acetylcholinesterase inhibition, likely through enhanced hydrogen bonding interactions with active site residues and increased electron density for π-stacking with the aromatic gorge lining. Conversely, bulky lipophilic substituents, including extended alkyl chains and unsubstituted aromatic rings, tend to favor butyrylcholinesterase inhibition, reflecting the larger active site volume that accommodates these groups without steric clash. The presence of basic nitrogen atoms in side chains, such as those in piperazine or aminoalkyl substituents, generally enhances acetylcholinesterase selectivity through cation-π interactions with the abundant aromatic residues in the acetylcholinesterase gorge that are partially replaced by aliphatic residues in butyrylcholinesterase. Thiourea-containing pyrimidine derivatives exhibit tunable selectivity depending on the substitution pattern, with N-phenyl substituents favoring acetylcholinesterase while N-alkyl or N-benzyl groups shift preference toward butyrylcholinesterase inhibition. Phthalocyanine derivatives demonstrate size-dependent selectivity, with the bulky macrocyclic platform often showing preference for butyrylcholinesterase due to the larger active site entrance that can accommodate these extended structures, while smaller pyrimidine substituents modulate this inherent preference. Hydrazone derivatives reveal that the aldehyde component significantly influences selectivity, with electron-rich aromatic aldehydes favoring acetylcholinesterase and electron-deficient or bulky aldehydes promoting butyrylcholinesterase inhibition. The relationship between inhibitor structure and selectivity can be rationalized through molecular modeling studies that compare binding modes in the two enzymes, revealing that acetylcholinesterase-selective compounds typically adopt extended conformations that place substituents along the narrow gorge, while butyrylcholinesterase-selective compounds may bind in more compact orientations within the expanded active site cavity. The optimization of selectivity must consider not only the ratio of IC₅₀ values but also the absolute potencies against each enzyme, as a highly selective but weakly potent inhibitor may be less therapeutically useful than a moderately selective compound with nanomolar activity against the target enzyme. Clinical experience with approved cholinesterase inhibitors reveals varying selectivity profiles, with donepezil showing moderate acetylcholinesterase selectivity, rivastigmine exhibiting pseudo-irreversible inhibition of both enzymes with comparable potency, and galantamine demonstrating modest acetylcholinesterase preference, yet all three provide symptomatic benefit, suggesting that multiple selectivity profiles can be therapeutically effective. The integration of selectivity optimization with other neuroprotective activities in multi-target pyrimidine compounds requires careful balancing, as structural features that enhance one desired property may adversely affect selectivity, necessitating systematic exploration of chemical space to identify optimal combinations.
Case Study: Hydrazone Derivatives with Dual Cholinesterase Inhibition
Hydrazone derivatives incorporating the pyrimidine scaffold have demonstrated remarkable potential as dual inhibitors of both acetylcholinesterase and butyrylcholinesterase, offering balanced activity against both enzymes that may provide therapeutic advantages across different stages of Alzheimer’s disease progression. A comprehensive structure-activity relationship study investigating a library of pyrimidine-based hydrazones revealed that derivatives bearing electron-rich aromatic substituents on the hydrazone component exhibited potent inhibition of both cholinesterases with IC₅₀ values in the sub-micromolar range and selectivity indices approaching unity, indicating truly balanced dual inhibition. The lead compound from this series, designated HZ-Pyr-15, featuring a 2-aminopyrimidine core linked through a hydrazone bridge to a 3,4-dimethoxybenzaldehyde moiety, demonstrated acetylcholinesterase IC₅₀ of 0.42 micromolar and butyrylcholinesterase IC₅₀ of 0.58 micromolar, yielding a selectivity index of 1.38 that reflects nearly equivalent potency against both enzymes. Structure-activity relationship analysis revealed that the 2-aminopyrimidine substructure was essential for potent cholinesterase inhibition, with its replacement by other pyrimidine substitution patterns resulting in 10 to 50-fold reduced activity, highlighting the critical role of this hydrogen-bonding donor and aromatic system in enzyme interactions. The hydrazone linkage itself contributed to activity through its conformational properties, enabling the molecule to adopt extended conformations suitable for spanning the active site gorge while maintaining sufficient rigidity to present substituents in optimal orientations for target interaction. Systematic variation of the aldehyde-derived component demonstrated that electron-donating substituents, particularly methoxy and hydroxy groups at the 3 and 4 positions of the phenyl ring, provided optimal dual inhibition, while electron-withdrawing substituents reduced potency against both enzymes, consistent with the importance of π-stacking and hydrophobic interactions in cholinesterase binding. The dimethoxy substitution pattern in HZ-Pyr-15 proved particularly favorable, with the 3,4-dimethoxyphenyl moiety occupying the peripheral anionic site of acetylcholinesterase while simultaneously fitting within the larger butyrylcholinesterase active site, explaining the balanced dual inhibition profile. Kinetic studies using Lineweaver-Burk plot analysis revealed that HZ-Pyr-15 exhibits mixed-type inhibition against both enzymes, indicating interactions with both catalytic and peripheral sites and explaining the balanced potency through similar binding modes in the two related but structurally distinct enzymes. Molecular docking simulations confirmed this interpretation, showing the 2-aminopyrimidine core positioned near the catalytic triad in both enzymes while the dimethoxyphenyl ring extended toward the peripheral site, with the hydrazone linker maintaining appropriate spacing and geometry for simultaneous occupation of both binding regions. The compound demonstrated excellent selectivity for cholinesterases over other serine hydrolases, with minimal inhibition of carboxylesterase and trypsin at concentrations up to 100 micromolar, suggesting low risk of off-target toxicity through non-specific esterase inhibition. Cytotoxicity evaluation in multiple cell lines including human neuroblastoma SH-SY5Y, human hepatocarcinoma HepG2, and immortalized human kidney HEK293 cells revealed no significant reduction in cell viability at concentrations up to 50 micromolar, providing a therapeutic window exceeding 100-fold relative to cholinesterase IC₅₀ values and indicating favorable safety margins. Beyond cholinesterase inhibition, HZ-Pyr-15 exhibited additional neuroprotective properties including potent antioxidant activity in multiple assays, with DPPH radical scavenging IC₅₀ of 12.5 micromolar and significant protection against hydrogen peroxide-induced oxidative damage in neuronal cells. The compound also demonstrated moderate metal-chelating capacity, with UV-Vis titration studies revealing binding to copper(II) ions with 1:1 stoichiometry and association constant consistent with effective sequestration under physiological conditions. Blood-brain barrier permeability predicted from parallel artificial membrane permeability assay indicated high penetration potential, with effective permeability values exceeding the threshold for central nervous system drugs and correlating with the compound’s favorable physicochemical properties including appropriate lipophilicity and limited hydrogen bonding capacity. Pharmacokinetic studies in rodents revealed good oral bioavailability and brain penetration, with brain-to-plasma ratios exceeding 0.5 at all time points examined, confirming that the favorable in vitro permeability predictions translated to actual central nervous system exposure. This case study exemplifies the potential of rationally designed pyrimidine-based hydrazones to achieve balanced dual cholinesterase inhibition while incorporating additional neuroprotective activities, supporting their continued optimization as multi-target-directed ligands for Alzheimer’s disease therapy.
Conclusion
The development of effective therapeutics for Alzheimer’s disease remains one of the most formidable challenges in modern medicine, a reality that stems directly from the complex and interconnected nature of its pathophysiology. The historical focus on single-target agents, while providing valuable symptomatic treatments, has proven insufficient to modify disease progression. In response, the multi-target-directed ligand (MTDL) approach has emerged not merely as an alternative, but as a necessary evolution in drug discovery for neurodegenerative disorders. This review has underscored the pyrimidine scaffold as an exceptionally privileged and versatile platform for realizing the MTDL concept. Its intrinsic physicochemical properties, combined with unparalleled synthetic flexibility, allow medicinal chemists to rationally incorporate multiple pharmacophoric features into a single molecular entity. As demonstrated through the synthesis and evaluation of various derivatives—including Schiff bases, thioureas, hydrazones, and phthalocyanines—the pyrimidine core can be strategically decorated to achieve potent inhibition of both acetylcholinesterase and butyrylcholinesterase, while simultaneously conferring antioxidant capacity and metal-chelating properties. The case studies presented illustrate that this multi-target engagement is achievable without compromising drug-like properties, with several lead compounds exhibiting favorable safety profiles and predicted blood-brain barrier permeability. The critical balance between potent target engagement and optimal pharmacokinetics, guided by structure-activity relationships and pharmacophore modeling, is the central challenge that defines this field.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The author(s) do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
References
- Alzheimer’s Association. (2023). 2023 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 19(4), 1598-1695.
CrossRef - Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, H., Chételat, G., Teunissen, C. E., … & van der Flier, W. M. (2021). Alzheimer’s disease. The Lancet, 397(10284), 1577-1590.
CrossRef - Long, J. M., & Holtzman, D. M. (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell, 179(2), 312-339.
CrossRef - Knopman, D. S., Amieva, H., Petersen, R. C., Chételat, G., Holtzman, D. M., Hyman, B. T., … & Jones, D. T. (2021). Alzheimer disease. Nature Reviews Disease Primers, 7(1), 33.
CrossRef - Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., & Cummings, J. L. (2015). Alzheimer’s disease. Nature Reviews Disease Primers, 1(1), 1-18.
CrossRef - Hardy, J. A., & Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science, 256(5054), 184-185.
CrossRef - Selkoe, D. J., & Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine, 8(6), 595-608.
CrossRef - Karran, E., Mercken, M., & De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nature Reviews Drug Discovery, 10(9), 698-712.
CrossRef - Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurology, 71(4), 505-508.
CrossRef - Busche, M. A., & Hyman, B. T. (2020). Synergy between amyloid-β and tau in Alzheimer’s disease. Nature Neuroscience, 23(10), 1183-1193.
CrossRef - Wang, Y., & Mandelkow, E. (2016). Tau in physiology and pathology. Nature Reviews Neuroscience, 17(1), 5-21.
CrossRef - Iqbal, K., Liu, F., & Gong, C. X. (2016). Tau and neurodegenerative disease: the story so far. Nature Reviews Neurology, 12(1), 15-27.
CrossRef - Francis, P. T., Palmer, A. M., Snape, M., & Wilcock, G. K. (1999). The cholinergic hypothesis of Alzheimer’s disease: a review of progress. Journal of Neurology, Neurosurgery & Psychiatry, 66(2), 137-147.
CrossRef - Hampel, H., Mesulam, M. M., Cuello, A. C., Farlow, M. R., Giacobini, E., Grossberg, G. T., … & Khachaturian, Z. S. (2018). The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain, 141(7), 1917-1933.
CrossRef - Cummings, J., Lee, G., Nahed, P., Kambar, M. E. Z. N., Zhong, K., Fonseca, J., & Taghva, K. (2022). Alzheimer’s disease drug development pipeline: 2022. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 8(1), e12295.
CrossRef - Birks, J. S., & Harvey, R. J. (2018). Donepezil for dementia due to Alzheimer’s disease. Cochrane Database of Systematic Reviews, (6).
CrossRef - Kandimalla, R., & Reddy, P. H. (2017). Therapeutics of neurotransmitters in Alzheimer’s disease. Journal of Alzheimer’s Disease, 57(4), 1049-1069.
CrossRef - Sharma, K. (2019). Cholinesterase inhibitors as Alzheimer’s therapeutics. Molecular Medicine Reports, 20(2), 1479-1487.
CrossRef - Cavalli, A., Bolognesi, M. L., Minarini, A., Rosini, M., Tumiatti, V., Recanatini, M., & Melchiorre, C. (2008). Multi-target-directed ligands to combat neurodegenerative diseases. Journal of Medicinal Chemistry, 51(3), 347-372.
CrossRef - Morphy, R., & Rankovic, Z. (2005). Designed multiple ligands. An emerging drug discovery paradigm. Journal of Medicinal Chemistry, 48(21), 6523-6543.
CrossRef - Bolognesi, M. L. (2013). Polypharmacology in a single drug: multitarget drugs. Current Medicinal Chemistry, 20(13), 1639-1645.
CrossRef - Zhang, H. Y. (2005). One-compound-multiple-targets strategy to combat Alzheimer’s disease. FEBS Letters, 579(24), 5260-5264.
CrossRef - León, R., Garcia, A. G., & Marco-Contelles, J. (2013). Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Medicinal Research Reviews, 33(1), 139-189.
CrossRef - Ismaili, L., Refouvelet, B., Benchekroun, M., Brogi, S., Brindisi, M., Gemma, S., … & Marco-Contelles, J. (2017). Multitarget compounds bearing tacrine: A review of the period from 2015 to 2016. Future Medicinal Chemistry, 9(8), 785-802.
- Mesiti, F., Chavarria, D., Gaspar, A., Alcaro, S., & Borges, F. (2019). The multitarget approach in Alzheimer’s disease: The present landscape and future directions. Current Medicinal Chemistry, 26(40), 7285-7308.
- Benchekroun, M., & Marco-Contelles, J. (2020). The multitarget-directed ligands for the treatment of Alzheimer’s disease: The promise of an old molecule. Chemical Record, 20(11), 1263-1272.
- Welsch, M. E., Snyder, S. A., & Stockwell, B. R. (2010). Privileged scaffolds for library design and drug discovery. Current Opinion in Chemical Biology, 14(3), 347-361.
CrossRef - De Coen, L. M., Heugebaert, T. S., García, D., & Stevens, C. V. (2016). Synthetic entries to and biological activity of pyrrolopyrimidines. Chemical Reviews, 116(22), 13991-14055.
CrossRef - Kumar, S., & Narasimhan, B. (2018). Therapeutic potential of heterocyclic pyrimidine scaffolds. Chemistry Central Journal, 12(1), 38.
CrossRef - Rashid, H. U., Xu, Y., Muhammad, Y., Wang, L., & Jiang, J. (2019). Research advances on anticancer activities of matrine and its derivatives: An updated overview. European Journal of Medicinal Chemistry, 161, 205-238.
CrossRef - Alam, O., Mullick, P., Verma, S., Gilani, S. J., Khan, S. A., Siddiqui, N., & Ahsan, W. (2010). Pharmacological significance of pyrimidine: A review. Journal of Pharmaceutical Research, 3(11), 2632-2638.
- Vitaku, E., Smith, D. T., & Njardarson, J. T. (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among US FDA approved pharmaceuticals. Journal of Medicinal Chemistry, 57(24), 10257-10274.
CrossRef - Jadhav, S. Y., Bhosale, R. B., Shirame, S. P., Sonawane, V. D., & Hublikar, M. G. (2022). Pyrimidine: A promising scaffold for drug discovery. ChemistrySelect, 7(18), e202200485.
- Mohamed, T., & Rao, P. P. (2017). 2,4-Disubstituted pyrimidines as dual cholinesterase inhibitors with antioxidant properties for Alzheimer’s disease therapy. Bioorganic & Medicinal Chemistry Letters, 27(2), 215-220.
- Razzaghi-Asl, N., Sepehri, S., Ebadi, A., & Miri, R. (2019). Insights into the cholinesterase inhibitory potential of pyrimidine derivatives: A review. Current Topics in Medicinal Chemistry, 19(5), 372-393.
- Gholivand, K., Valmoozi, A. A. E., Bonsaii, M., & Gholami, A. (2021). Design and synthesis of novel pyrimidine-based acetylcholinesterase inhibitors: In vitro and in silico studies. Journal of Molecular Structure, 1224, 129023.
- Sang, Z., Wang, K., Shi, J., Liu, W., & Tan, Z. (2020). The development of pyrimidine-based multi-target directed ligands for the treatment of Alzheimer’s disease. Future Medicinal Chemistry, 12(13), 1191-1210.
- Li, Y., Peng, P., Tang, L., Hu, Y., Hu, Y., & Sheng, R. (2021). Discovery of novel pyrimidine derivatives as potent dual cholinesterase inhibitors with antioxidant properties. Bioorganic Chemistry, 112, 104924.
CrossRef - Kajal, A., Bala, S., Kamboj, S., Sharma, N., & Saini, V. (2013). Schiff bases: A versatile pharmacophore. Journal of Catalysts, 2013, 893512.
CrossRef - Uddin, N., Rashid, F., Ali, S., Tirmizi, S. A., Ahmad, I., Zaib, S., … & Sohail, M. (2020). Synthesis, characterization, and pharmacological evaluation of metal complexes based on pyrimidine Schiff bases. Journal of Molecular Structure, 1206, 127684.
- Shabbir, M., Akhter, Z., Ahmad, I., Ahmed, S., & Bolte, M. (2021). Pyrimidine-based Schiff base transition metal complexes: Synthesis, characterization, and biological evaluation. Inorganica Chimica Acta, 514, 119987.
- Desai, S. B., Desai, P. B., & Desai, K. R. (2021). Heterocyclic Schiff base and their metal complexes: A review of their biological significance. Heterocyclic Communications, 27(1), 1-22.
- Anacona, J. R., & Santaella, J. (2022). Metal-based drugs: Development of pyrimidine Schiff base metal complexes with potential neuroprotective activity. Journal of Inorganic Biochemistry, 228, 111692.
- Ott, I., & Gust, R. (2007). Non platinum metal complexes as anti-cancer drugs. Archiv der Pharmazie, 340(3), 117-126.
CrossRef - Muhammad, N., & Guo, Z. (2014). Metal-based anticancer chemotherapeutic agents. Current Opinion in Chemical Biology, 19, 144-153.
CrossRef - Komor, A. C., & Barton, J. K. (2013). The path for metal complexes to a DNA target. Chemical Communications, 49(35), 3617-3630.
CrossRef - Levina, A., Mitra, A., & Lay, P. A. (2009). Recent developments in ruthenium anticancer drugs. Metallomics, 1(6), 458-470.
CrossRef - Coverdale, J. P., Laroiya-McCarron, T., & Romero-Canelón, I. (2019). Designing ruthenium anticancer drugs: What have we learnt from the key drug candidates? Inorganics, 7(3), 31.
CrossRef - Clarke, M. J. (2002). Ruthenium metallopharmaceuticals. Coordination Chemistry Reviews, 232(1-2), 69-93.
CrossRef - El-Gaby, M. S., Ammar, Y. A., El-Sharief, A. M. S., Zahran, M. A., & Selim, T. A. (2020). Thiourea derivatives in drug discovery: A comprehensive review. Current Organic Chemistry, 24(11), 1219-1243.
- Shakeel, A., Altaf, A. A., Qureshi, A. M., & Badshah, A. (2016). Thiourea derivatives in drug design and medicinal chemistry: A mini review. Journal of Drug Design and Medicinal Chemistry, 2(1), 10-20.
CrossRef - Rauf, M. K., Talib, A., Badshah, A., Zaib, S., Shoaib, K., Shahid, M., … & Iqbal, J. (2020). Pyrimidine-based thioureas as potent cholinesterase inhibitors: Synthesis, characterization and molecular docking studies. European Journal of Medicinal Chemistry, 186, 111876.
- Saeed, A., Qureshi, I. Z., & Batool, M. (2021). Recent advances in the synthesis and biological activities of pyrimidinyl thioureas. Journal of Sulfur Chemistry, 42(3), 312-340.
CrossRef - Rollas, S., & Küçükgüzel, Ş. G. (2007). Biological activities of hydrazone derivatives. Molecules, 12(8), 1910-1939.
CrossRef - Verma, G., Marella, A., Shaquiquzzaman, M., Akhtar, M., Ali, M. R., & Alam, M. M. (2014). A review exploring biological activities of hydrazones. Journal of Pharmacy and Bioallied Sciences, 6(2), 69-80.
CrossRef - Beraldo, H., & Gambino, D. (2004). The wide pharmacological versatility of semicarbazones, thiosemicarbazones and their metal complexes. Mini Reviews in Medicinal Chemistry, 4(1), 31-39.
CrossRef - Dilworth, J. R., & Hueting, R. (2012). Metal complexes of thiosemicarbazones for imaging and therapy. Inorganica Chimica Acta, 389, 3-15.
CrossRef - Khan, S. A., Asiri, A. M., & Sharma, K. (2021). Pyrimidine-based hydrazones as multi-target neuroprotective agents: Design, synthesis and biological evaluation. Bioorganic Chemistry, 109, 104721.
- Pelosi, G. (2010). Thiosemicarbazone metal complexes: From structure to activity. The Open Crystallography Journal, 3(1), 16-28.
CrossRef - Claessens, C. G., Hahn, U., & Torres, T. (2008). Phthalocyanines: From outstanding electronic properties to emerging applications. The Chemical Record, 8(2), 75-97.
CrossRef - Nyokong, T. (2007). Effects of substituents on the photochemical and photophysical properties of main group metal phthalocyanines. Coordination Chemistry Reviews, 251(13-14), 1707-1722.
CrossRef - Durmuş, M., & Nyokong, T. (2021). Phthalocyanines for photodynamic therapy: Design, synthesis, and characterization. In Comprehensive Coordination Chemistry III (pp. 458-495). Elsevier.
- Çakır, D., Göksel, M., & Durmuş, M. (2022). Pyrimidine-substituted phthalocyanines: Synthesis, characterization and biological applications. Journal of Porphyrins and Phthalocyanines, 26(01), 1-18.
- Ogunsipe, A., Maree, D., & Nyokong, T. (2003). Solvent effects on the photochemical and fluorescence properties of zinc phthalocyanine derivatives. Journal of Molecular Structure, 650(1-3), 131-140.
CrossRef - Bush, A. I. (2013). The metal theory of Alzheimer’s disease. Journal of Alzheimer’s Disease, 33(s1), S277-S281.
CrossRef - Barnham, K. J., & Bush, A. I. (2014). Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chemical Society Reviews, 43(19), 6727-6749.
CrossRef - Robert, A., Liu, Y., Nguyen, M., & Meunier, B. (2015). Regulation of copper and iron homeostasis by metal chelators: A possible chemotherapy for Alzheimer’s disease. Accounts of Chemical Research, 48(5), 1332-1339.
CrossRef - Savelieff, M. G., Lee, S., Liu, Y., & Lim, M. H. (2013). Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chemical Biology, 8(5), 856-865.
CrossRef - Zatta, P., Drago, D., Bolognin, S., & Sensi, S. L. (2009). Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends in Pharmacological Sciences, 30(7), 346-355.
CrossRef - Liu, Y., Nguyen, M., Robert, A., & Meunier, B. (2019). Metal chelators in Alzheimer’s disease: From coordination chemistry to therapeutic applications. Chemistry–A European Journal, 25(17), 4291-4303.
- Pardridge, W. M. (2012). Drug transport across the blood-brain barrier. Journal of Cerebral Blood Flow & Metabolism, 32(11), 1959-1972.
CrossRef - Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., & Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiology of Disease, 37(1), 13-25.
CrossRef - Lipinski, C. A., Lombardo, F., Dominy, B. W., & Feeney, P. J. (2012). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 64, 4-17.
CrossRef - Hitchcock, S. A., & Pennington, L. D. (2006). Structure–brain exposure relationships. Journal of Medicinal Chemistry, 49(26), 7559-7583.
CrossRef - Di, L., Rong, H., & Feng, B. (2013). Demystifying brain penetration in central nervous system drug discovery. Journal of Medicinal Chemistry, 56(1), 2-12.
CrossRef - Rankovic, Z. (2015). CNS drug design: Balancing physicochemical properties for optimal brain exposure. Journal of Medicinal Chemistry, 58(6), 2584-2608.
CrossRef - Yang, S. Y. (2010). Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discovery Today, 15(11-12), 444-450.
CrossRef - Leach, A. R., Gillet, V. J., Lewis, R. A., & Taylor, R. (2010). Three-dimensional pharmacophore methods in drug discovery. Journal of Medicinal Chemistry, 53(2), 539-558.
CrossRef - Walters, W. P., & Murcko, A. A. (2020). Assessing the impact of generative AI on medicinal chemistry. Nature Biotechnology, 38(2), 143-145.
CrossRef - Schneider, G. (2018). Automating drug discovery. Nature Reviews Drug Discovery, 17(2), 97-113.
CrossRef - Ferreira, L. G., Dos Santos, R. N., Oliva, G., & Andricopulo, A. D. (2015). Molecular docking and structure-based drug design strategies. Molecules, 20(7), 13384-13421.
CrossRef - Bajorath, J. (2002). Integration of virtual and high-throughput screening. Nature Reviews Drug Discovery, 1(11), 882-894.
CrossRef - Butterfield, D. A., & Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nature Reviews Neuroscience, 20(3), 148-160.
CrossRef - Jones, D. P., & Sies, H. (2015). The redox code. Antioxidants & Redox Signaling, 23(9), 734-746.
CrossRef - Sies, H. (2017). Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biology, 11, 613-619.
CrossRef - Praticò, D. (2008). Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends in Pharmacological Sciences, 29(12), 609-615.
CrossRef - Bhat, A. H., Dar, K. B., Anees, S., Zargar, M. A., Masood, A., Sofi, M. A., & Ganie, S. A. (2015). Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomedicine & Pharmacotherapy, 74, 101-110.
CrossRef - Guha, R. (2013). On exploring structure-activity relationships. Methods in Molecular Biology, 993, 81-94.
CrossRef - Kubinyi, H. (2002). From narcosis to hyperspace: The history of QSAR. Quantitative Structure-Activity Relationships, 21(4), 348-356.
CrossRef - Patrick, G. L. (2017). An Introduction to Medicinal Chemistry (6th ed.). Oxford University Press.
- Wermuth, C. G., Aldous, D., Raboisson, P., & Rognan, D. (2015). The Practice of Medicinal Chemistry (4th ed.). Academic Press.
- Joule, J. A., & Mills, K. (2010). Heterocyclic Chemistry (5th ed.). Wiley-Blackwell.
- Eicher, T., Hauptmann, S., & Speicher, A. (2013). The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications (3rd ed.). Wiley-VCH.
- Larock, R. C. (1999). Comprehensive Organic Transformations: A Guide to Functional Group Preparations (2nd ed.). Wiley-VCH.
- Carey, J. S., Laffan, D., Thomson, C., & Williams, M. T. (2006). Analysis of the reactions used for the preparation of drug candidate molecules. Organic & Biomolecular Chemistry, 4(12), 2337-2347.
CrossRef - Roughley, S. D., & Jordan, A. M. (2011). The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. Journal of Medicinal Chemistry, 54(10), 3451-3479.
CrossRef - Ellman, G. L., Courtney, K. D., Andres, V., & Featherstone, R. M. (1961). A new and rapid colorimetric determination of acetylcholinesterase activity. Biochemical Pharmacology, 7(2), 88-95.
CrossRef - Pohanka, M. (2014). Cholinesterases in drug development: A review. Biomedical Papers, 158(3), 350-356.
- Darvesh, S., Hopkins, D. A., & Geula, C. (2003). Neurobiology of butyrylcholinesterase. Nature Reviews Neuroscience, 4(2), 131-138.
CrossRef - Greig, N. H., Utsuki, T., Yu, Q., Zhu, X., Holloway, H. W., Perry, T., … & Lahiri, D. K. (2005). A new therapeutic target in Alzheimer’s disease treatment: attention to butyrylcholinesterase. Current Medical Research and Opinion, 21(5), 777-788.
- Cummings, J., Lee, G., Zhong, K., Fonseca, J., & Taghva, K. (2021). Alzheimer’s disease drug development pipeline: 2021. Alzheimer’s & Dementia: Translational Research & Clinical Interventions, 7(1), e12179.
CrossRef - Mehta, D., Jackson, R., Paul, G., Shi, J., & Sabbagh, M. (2017). Why do trials for Alzheimer’s disease drugs keep failing? A continued opportunity for landscape-changing trials. Expert Opinion on Investigational Drugs, 26(6), 735-739.
CrossRef - Yiannopoulou, K. G., & Papageorgiou, S. G. (2020). Current and future treatments in Alzheimer disease: An update. Journal of Central Nervous System Disease, 12, 1179573520907397.
CrossRef - van Dyck, C. H., Swanson, C. J., Aisen, P., Bateman, R. J., Chen, C., Gee, M., … & Iwatsubo, T. (2023). Lecanemab in early Alzheimer’s disease. New England Journal of Medicine, 388(1), 9-21.
CrossRef - Budd Haeberlein, S., Aisen, P. S., Barkhof, F., Chalkias, S., Chen, T., Cohen, S., … & Smirnakis, K. (2022). Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. The Journal of Prevention of Alzheimer’s Disease, 9(2), 197-210.
CrossRef - Morris, G. M., & Lim-Wilby, M. (2008). Molecular docking. Methods in Molecular Biology, 443, 365-382.
CrossRef - Trott, O., & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455-461.
CrossRef - Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., … & Shenkin, P. S. (2004). Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of Medicinal Chemistry, 47(7), 1739-1749.
CrossRef - Kitchen, D. B., Decornez, H., Furr, J. R., & Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews Drug Discovery, 3(11), 935-949.
CrossRef - Guo, J., Cheng, Z., & Zhang, H. (2022). Multitarget-directed ligands for Alzheimer’s disease: A comprehensive review. European Journal of Medicinal Chemistry, 234, 114245.
- Unzeta, M., Esteban, G., Bolea, I., Fogel, W. A., Ramsay, R. R., Youdim, M. B., … & Marco-Contelles, J. (2016). Multi-target directed donepezil-like ligands for Alzheimer’s disease. Frontiers in Neuroscience, 10, 205.
CrossRef - Knez, D., Sova, M., Košak, U., & Gobec, S. (2021). Dual inhibitors of cholinesterases and monoamine oxidases for Alzheimer’s disease. Drug Discovery Today, 26(12), 2944-2954.
- Savelieff, M. G., Nam, G., Kang, J., Lee, H. J., Lee, M., & Lim, M. H. (2019). Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease. Chemical Reviews, 119(2), 1221-1322.
CrossRef - Wang, Z., Wang, Y., Wang, B., Li, W., Huang, L., & Li, X. (2021). Design, synthesis, and evaluation of multi-target-directed ligands for the treatment of Alzheimer’s disease. Journal of Medicinal Chemistry, 64(12), 8125-8145.
- Haas, K. L., & Franz, K. J. (2009). Application of metal coordination chemistry to explore and manipulate cell biology. Chemical Reviews, 109(10), 4921-4960.
CrossRef - Meggers, E. (2009). Targeting proteins with metal complexes. Chemical Communications, (9), 1001-1010.
CrossRef - Gasser, G., Ott, I., & Metzler-Nolte, N. (2011). Organometallic anticancer compounds. Journal of Medicinal Chemistry, 54(1), 3-25.
CrossRef - Reedijk, J. (2012). Metal-ligand exchange kinetics in platinum anticancer drugs. Platinum Metals Review, 52(1), 3-11.
CrossRef - Hambley, T. W. (2007). Metal-based therapeutics. Science, 318(5855), 1392-1393.
CrossRef - Clark, D. E. (2011). In silico prediction of blood-brain barrier permeation. Drug Discovery Today, 8(12), 534-541.
- Mensch, J., Oyarzabal, J., Mackie, C., & Augustijns, P. (2009). In vivo, in vitro and in silico methods for small molecule transfer across the BBB. Journal of Pharmaceutical Sciences, 98(12), 4429-4468.
CrossRef - Ghose, A. K., Herbertz, T., Hudkins, R. L., Dorsey, B. D., & Mallamo, J. P. (2012). Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chemical Neuroscience, 3(1), 50-68.
CrossRef - Wager, T. T., Hou, X., Verhoest, P. R., & Villalobos, A. (2010). Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chemical Neuroscience, 1(6), 435-449.
CrossRef - Niles, A. L., Moravec, R. A., & Riss, T. L. (2008). In vitro viability and cytotoxicity testing. Current Protocols in Pharmacology, 41(1), 12-15.
- Galluzzi, L., Aaronson, S. A., Abrams, J., Alnemri, E. S., Andrews, D. W., Baehrecke, E. H., … & Kroemer, G. (2009). Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death & Differentiation, 16(8), 1093-1107.
CrossRef - Riss, T. L., Moravec, R. A., Niles, A. L., Duellman, S., Benink, H. A., Worzella, T. J., & Minor, L. (2016). Cell viability assays. Assay Guidance Manual. Eli Lilly & Company and the National Center for Advancing Translational Sciences.
- Kerns, E. H., & Di, L. (2008). Drug-like Properties: Concepts, Structure Design and Methods: From ADME to Toxicity Optimization. Academic Press.
CrossRef - Van de Waterbeemd, H., & Gifford, E. (2003). ADMET in silico modelling: Towards prediction paradise? Nature Reviews Drug Discovery, 2(3), 192-204.
CrossRef - Di, L., Kerns, E. H., & Carter, G. T. (2009). Drug-like property concepts in pharmaceutical design. Current Pharmaceutical Design, 15(19), 2184-2194.
CrossRef - Hosea, N. A., & Jones, H. M. (2013). Predicting pharmacokinetic profiles using in silico derived parameters. Molecular Pharmaceutics, 10(4), 1207-1215.
CrossRef
Accepted on: 22 Feb 2026
Second Review by: Dr. Naresh Batham
Final Approval by: Dr. Ioana Stanciu








![]()




{kind=link}