A Comparative Study of Binding of Different Drugs on gp120: Insight from Molecular Dynamics Simulation Study
Vishnudatt Pandey , Gargi Tiwari and Rajendra Prasad Ojha
, Gargi Tiwari and Rajendra Prasad Ojha
Biophysics Unit, Department of Physics, Deen Dayal Upadhyay Gorakhpur University, Gorakhpur, India.
Corresponding Author E-mail: vishnudattpandey127@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/340635
Article Received on : 15-10-2018
Article Accepted on : 26-11-2018
Article Published : 23 Nov 2018
HIV-I cellular infection triggered by CD4 receptor protein and viral envelop glycoprotein gp120 binding event. CD4:gp120 surface is directed by the contact points of a hydrophobic gp120 cavity capped by Phe43CD4 and ionic bonds residues Arg59CD4 and Asp368gp120. The binding sites originated by gp120 and CD4 interaction leads to the entry of HIV-I into the host membrane, where, gp120 and a CD4 binding site becomes the main mark for plenty of drug uncovering program. Here, we took the crystal structure of small-molecule of gp120 in a complex that concurrently pursues both of the hotspots of gp120 binding sites. All ligands in our study are small molecules that are able to obstruct the protein-protein interactions between CD4 and gp120. This study aims at the thermodynamical insights of the ligand binding in CD4 binding sites using Molecular Dynamics Simulations Study and calculation of binding free energy. The physical of binding of drugs distinctly indicates a hydrophobic and electrostatics interaction motivated binding of ligands which explicitly mark CD4 binding sites.
KEYWORDS:Gp120 Binding; HIV-1 Entry; MD Simulations; MMGBSA
Download this article as:| Copy the following to cite this article: Pandey V, Tiwari G, Ojha R. P. A Comparative Study of Binding of Different Drugs on gp120: Insight from Molecular Dynamics Simulation Study. Orient J Chem 2018;34(6). |
| Copy the following to cite this URL: Pandey V, Tiwari G, Ojha R. P. A Comparative Study of Binding of Different Drugs on gp120: Insight from Molecular Dynamics Simulation Study. Orient J Chem 2018;34(6). Available from: http://www.orientjchem.org/?p=53053 |
Introduction
By the end of 2017, over 70 million people were infected by HIV-1/AIDS, and about 36.9 million people are still septic by HIV-1.1-3 HIV-1(Virus type-I) changes in cells, as an originator gp160, and afterwards, split to go-41 and mature gp120. The HIV-1 Env (Envelope protein), target host cells on viral entry into the cells,4-6 that mechanized as a trimer of gp120-gp 41. Receptor CD4 and its co-receptor CCR5/CXCR4 binds with Env-gp120, in an indirect virus-cell membrane fusion. Viral Env protein undergoes big conformational alteration during its entry process. CD4 induces conformational changes into gp120 upon its binding, thus facilitating subsequent interaction through the CCR5 or CXR4 co-receptors. Gp120 binds together with receptor CD4 and co-receptor CCR5/CXCR4, therefore, triggering an extreme conformational change in gp41 which permits viral and cell membranes fusions. HIV pathological process is aided by a number of attachment effect whose initiation is carried out by the viral envelope glycoproteins. These glycoproteins are organized into trimeric spikes which are present on the surface of the virion.7 Every individual trimeric envelope confounds consist of three gp 41 trans membrane proteins and three gp120 envelope glycoproteins.8 Each of these processes, in fact, the overall step informs new potent mark for drug discovery.9-11 Apparently, initial entry step of the virion into CD4 cells is an important yet difficult approach for the prevention of HIV-1 pathological process and AIDS. Indeed, there have not been thorough studies on designing viral entry drugs that mark this entry process.12 At the CD4−gp120 interaction point, residue Phe43 of CD4 is situated on the CDR2-like loop. This residue binds inside the hydrophobic cavity of gp120, this cavity known as “Phe43 cavity”, whereas Arg59 of CD4 is situated on a neighbour β-strand and creates electrostatic interaction with Asp368 of gp120.13-14 Debnath et. al, recognized two drugs of CD4−gp120 namely NBD-556 and NBD-55715 by preceding transmission of small-molecule inhibitors of viral fusion. Dual Hotspot HIV-1 Entry Inhibitors which engage both the sites i.e. hydrophobic Phe43 cavity and electrostatic interaction with Asp368 of gp120 with Arg59 of CD4, are used for inhibition.16
Although important residues are active in protein-ligand complexes, as suggested by present structural information, they do not furnish physical knowledge for the prominence in interaction pair of each residue. Hence, these become mandatory to clarify the physical assistance of phenomenon at the level of atomic resolution, so as to double-check the physical assistance of all residue near the attraction site participating in the stabilization of the complex. Here we performed computational methods to investigate the interaction between gp120 and ligands using MD simulation methods. Our findings promote us to validate the thermodynamic result. In addition, this analysis shows that the method used is MMGBSA method and is capable to imitate the experimental validation of the result of binding free energies.
Materials and Methods
The initial coordinates of the gp120- ligand complexes were obtained from the Protein Data Bank (PDB). The following PDB entries were used to construct our models: gp120 complex(gp120-0LM) with N-(4-chloro-3- fluorophenyl)-N’-(1,2,2,6,6-pentamethylpiperidin-4-yl) ethanediamide (0LM) code 4DKO; gp120 complex (gp120- 0LL) with N-[(1S,2S)-2-amino-2,3-dihydro-1H-inden-1-yl]-N’-(4-chloro-3-fluorophenyl) ethanediamide (0LL), code 4DKP; gp120 complex (gp120-0LK) with N-[(1S,2S)-2-carbamimidamido-2,3-dihydro-1H-inden-1-yl]-N’-(4-chloro-3-fluorophenyl) ethanediamide (0LK), code 4DKQ; and gp120 complex (gp120-0LJ) with (N- [(1R,2R)-2-carbamimidamido-2,3-dihydro-1H-inden-1-yl]-N’-(4-chloro-3-fluorophenyl)ethanediamide (0LJ), code 4DKR.16 Chemical structure of all the ligand is shown in figure 1.
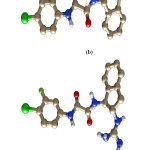 |
Figure 1: Chemical structure of all the inhibitors named as a) 0LM, b) 0LL, c) 0LK, d) 0LJ respectively from top to bottom. |
The coordinate and parameter of the hydrogen atom for gp120 were generated by using LEAP module of using ff12SB AMBER force filed.17 Optimization of all ligands was accomplished by HF (Hartree-Fock) methodology with 6-31G* basis set. Once the geometry optimization was done, consecutive frequencies were calculated to assure the fixed points. The partial atomic charge was calculated by Restrained electrostatic potential method (RESP).18-19 The complex was neutralized and solvated by using Na+ and TIP3P octahedral water-box.20 The solvated complex was then gradually strengthened from 10 to 300K for the period of 200 ps after that the system sustained in isothermal-isobaric(NPT) ensemble, to get the 300K temperature using Langevin thermostat21 and 1atm pressure by using Barendsen barostat22 with a collision frequency of 2ps and pressure relaxation time 1ps. SHAKE23 was used for constraining the hydrogen bonds. For treating long-range electrostatic, Particle Mesh Ewald (PME) method24 was used. Once the system attained a 300K temperature and 1atm pressure, the equilibrium dynamics was carried for 4ns, with the previously described parameters. Afterwards, production dynamics was started and continued up to 200 ns for the protein-ligand system. The coordinate construction in the trajectories of production dynamics was gathered at an interval of 10ps. Ptraj module of Amber14 was employed to carry out all the analysis of trajectories while for visualization of structure VMD 1.6.7,25 Chimera-1.526 module was used for the image purpose.
Calculation of Absolute Binding Free Energies and Per-Residue Calculation
For the calculations of thermodynamic parameters and free energy of binding, MM-GBSA method was used. The principles of these methods are all well constituted and have been taken up elsewhere.27-29 MMGBSA method used because it has been favourably applied for the analogous system, in this study but of various class in past studies.30-35
The specific parameters employed in our approach are described here. The binding free energy (ΔGmmgbsa) of the complex was calculated by using the following:
ΔGmmgbsa = Gcomplex -Greceptor -Gligand
ΔGbind = ΔEMM +ΔGGB +ΔGSA -TΔS
where ΔEMM is the total molecular mechanics energy of the molecular system in the gas phase, including the van der Waals (ΔEvdw) and electrostatic (ΔEele) interaction energies. ΔGsol and ΔGele, sol are electrostatic and non polar contributions to desolvation upon ligand binding, respectively, and -TΔS is the entropy contribution arising from changes in the degrees of freedom of the solute molecules, which we reconsidered here to obtain ΔGbind; therefore, our values reported for the MMGBSA calculations can be called absolute binding free energies. In order to get the crucial residue study, the absolute binding free energies were determined in terms of the contributions of each individual residues by using free-energy-per-residue decomposition theory.
Result
Binding Free Energy Calculation Binding free energy of ligand with the receptor was calculated by using snapshots collected from trajectories during the last 40 ns time of Molecular Dynamics trajectories when the RMSD converges figure 2(a )and the finding are listed in Table 1. Binding free energy for all complex is shown in figure 2(b). According to this result the binding free energy (ΔGbind) of the gp 120-0LM, gp 120-0LL, gp 120- 0LK, gp 120-0LJ complexes are − 8.46, − 6.23,− 12.67 and− 8.39 kcal mol−1, respectively, these results agree with the experimental findings. This finding discloses that the binding abilities of the third inhibitor, 0LK, are stronger than the other 3 inhibitors 0LL, 0LK and 0LJ. Moreover, the change in entropy (− TΔ S), induced by the inhibitor bindings yield a good correlation with enthalpy interaction(ΔHot). As observed from Table 1, the van der Waals interaction energy term (ΔEvdW), non-polar solvation energy term (ΔGnp) give satisfactory involvement with inhibitor binding. Although the inhibitor bindings are favoured by the electrostatic interaction (ΔEele), this favourable factor is completely regulated by stronger unfavourable polar solvation energy term (Δ Ggb).
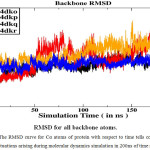 |
Figure 2a: The RMSD curve for Cα atoms of protein with respect to time tells conformational fluctuations arising during molecular dynamics simulation in 200ns of time scale. |
In contrast to the gp120-0LK complex, the van der Waals energy term and non-polar solvation energy term of the complex gp 120-0LM binding are decreased by 13.83, and 1.42 kcal mol−1, respectively. Similarly, in gp 120-0LL too, the binding leads to the reduction of van der Waals energy and non-polar solvation energy by 6.91 and 0.68 kcal mol−1 respectively. Similarly, in gp 120-0LJ the binding also goes to the reduction in van der Waals energy and non-polar solvation energy by 3.60 and 0.09 kcal mol−1 respectively. In a nutshell, this reduction in the van der Waals interaction may be considered as the main source of weaker binding affinities of inhibitors to 0LK, 0LL, and 0LJ in contrast to 0LK. The energetic contributions reveal that the association between gp120 and the four different.
 |
Figure 2b: Binding Energy for all the four complexes is shown in the figure. |
Standard deviations for all the energy terms are shown in table 1
ligands are chiefly governed by nonpolar energy (ΔEnonpolar), with which the van der Waals energy (ΔEvdw) contribute greatly. The gas-phase electrostatic energy (ΔEele) of the complexes are found to be favourable. For the first complex, gp 120-0LM, electrostatic energy are very low and it is quite evident that it does not make strong Hbond with inhibitor.
Table 1: Binding free energies (kcal•mol− 1). With Errors are written by signs “±” represents the standard errors of the mean.
| Ligand | ΔEele | ΔEvdw | ΔGnon–pol | ΔGGB | ΔHtot | -TΔS | ΔGexp | ΔGbind |
| 0LM | -6.19± 3.86 | -30.97± 3.97 | -3.95± 0.48 | 17.59± 3.42 | -23.53±3.7 | 15.0± 1.75 | -8.80 | -8.46 |
| 0LL | -40.59± 3.86 | -37.89± 2.58 | -4.69± 0.21 | 56.85± 10.20 | -26.32± 2.8 | 20.08± 3.9 | -7.90 | -6.23 |
| 0LK | -67.63± 1.02 | -44.80± 2.33 | -5.37± 0.15 | 79.88± 3.57 | -37.93± 3.9 | 25.25± 1.9 | -9.0 | -12.67 |
| 0LZ | -57.85±12.95 | -41.20± 1.95 | -5.58± 0.04 | 73.01± 8.37 | -31.62± 2.0 | -23.2± 3.9 | -8.90 | -8.39 |
Here:
ΔEvdw = van der Waals interaction term on ligand associations, ΔEele = electrostatic interaction term on ligand associations, ΔEpolar = polar interaction term on ligand associations,
ΔEnonpolar = nonpolar interaction term on ligand associations.
ΔGgas = ΔEVDW + ΔEEEL,
ΔGsolv = ΔEpolar + ΔEnonpolar,
ΔGbinding = ΔGgas + ΔGsolv
Scrutiny of the entropic contributions (TΔS) demonstrates that the formed complexes are distinguished by unfavourable entropy values due to a reduction in the degrees of freedom. The lowest entropy change (TΔS) was observed in the arrangement of the first complex gp120-0LM with ligand 0LM and the value of entropy change (TΔS) improved in terms of chain length thus disclosing that the ligand size plays a significant part in entropic part (Fig. 1). The Colour map for all the residues of the receptor is shown in Fig. 3. Here, acidic residues like aspartic acid and glutamic acid are shown in red, hydrophobic residues (Ala, Val, Ile, Leu, Tyr, Phe, Trp, Met, Cys, Pro) in white, basic residues like histidine, lysine, arginine in blue, polar residues (Ser, Thr, Gln, Asn) in green and other residues like glycine in gray.
 |
Figure 3: Colour map for all the residues. |
Residue-wise Decomposition Free Energy
Energy decomposition investigation allows us to explain the part of respective amino acid in deciding complex stabilization. The figure demonstrates the primal residues for bonding and the contributions of total free energy(ΔGtotal).
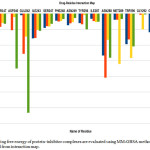 |
Figure 4: The binding free energy of protein–inhibitor complexes are evaluated using MM-GBSA methodology which is depicted from interaction map. |
These figures represent that all the complexes are stabilized mainly by hydrophobic and polar amino acid (Fig 4). All ligands are surrounded by several hydrophobic and polar residues and detail interaction of this amino acid with all the four drugs is shown in figure( Fig 5). It is self- evident that the ligands 0LM and 0LL interact with a maximum number of amino acids. In case of 0LM – Trp 290, Met 289, Asn 288, and Glu 242 play strongly and are principal amino acid responsible for strong binding. For 0LL – Asn 288, and Ile 338 provide the main interactions. Similar is the case with ligand 0LK in which Trp 290, Met 289, Asn 288, Ile243 and Glu 242 are crucial. Also, in regard to the ligand 0LZ, Trp 290, Asn 288, and Glu 242 are the major contributors for favourable interactions.
 |
Figure 5: Name of Residues who surrounded around all inhibitors for all complex 4DKO-0LM, 4DKP-0LL, 4DKQ-0LK and 4DKR-0LZ from top to bottom respectively. |
In accordance with the total free energy contributions, residues Trp 290, Asn 288 and Glu 242 of gp 120 have the greatest impact in the binding energy which proposes that these amino acid play a critical part in ligand binding. In addition, the role of polar, non-polar, van der Waals and electrostatics energy for all the amino acid are given in (table 2) for all the four complexes.
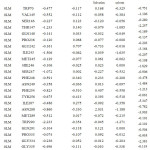 |
Table 2a: The contribution of each energy terms in binding affinity for complex named as 4DKO-0LM. |
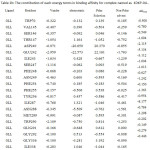 |
Table 2b: The contribution of each energy terms in binding affinity for complex named as 4DKP-0LL. |
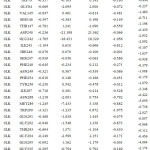 |
Table 2c: The contribution of each energy terms in binding affinity for complex named as 4DKQ-0LK.Click here to View table |
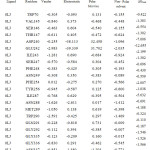 |
Table 2d: The contribution of each energy terms in binding affinity for complex named as 4DKR-0LZ. |
From figure one can infer that the Van der Walls and electrostatics energy play an essential role in total binding energy for almost all hydrophobic and polar residues. Asn288 and Glu242 have maximum electrostatics contribution in total binding affinity. Still, hydrophobic residues contribute via Van der Walls interaction.
Discussion
Here, the binding interaction between gp120 and four experimentally known ligands were evaluated with the help of conformational analysis and the binding free energy calculated over 200 ns dynamics using the MMGBSA methodology. The energetic analysis revealed a qualitative agreement of the theoretically calculated binding free energies with their experimentally reported values. The calculated results tell that the inhibitors produce stronger binding to 0LK as compared to 0LM,0LL, 0LZ. Also, the increase in van der Walls interaction of inhibitors with 0LK relative to the other inhibitors is the main factor responsible for stronger bindings of inhibitors. The fact that the reduction in Van der Waals energy may be a main cause of weaker binding of inhibitor 0LK than to other inhibitors.
However, decomposition free energy asserted not only that plausive free energies arise only from favourable interactions due to residues ASn288 and Glu242 but also these actions were amongst the most favourable role, proclaiming the essential contribution of these energy interactions is helpful in the ligand structure. The Entropic analysis demonstrated that all four complexes did undergo a conformational reduction which in turn played important role in bringing the MMGBSA results in more closer to the observational absolute binding free energies.
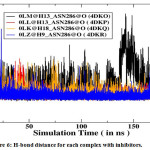 |
Figure 6: H-bond distance for each complex with inhibitors. |
Furthermore, energetic contributions to the binding are attributed by a large number of hydrophobic contacts. Asn288 makes a strong H-bond with all inhibitors and is significantly accountable for electrostatic interactions (Figure 5). Moreover, Trp290 also provides a great energetic interaction via its hydrophobic side chain and is the chief contributor for the enhanced van der Waals interactions.
Conclusion
In the present study, the per-residue binding free energy decomposition tells us to acknowledge Asn288, Glu242, Trp290 Asp240 and Met289 as the crucial amino acid for the complex stabilization of the four ligands, which is also in good agreement with experimental values for the GP120 complexes. Val145, Thr147, Glu242, Ser247, Tyr256, Ile287, Met289, Gly292, Gly335, Asn337, and Ile338 as the chief amino acid for the complex stabilization of the four ligands. Since these residues are very close and near to the binding site, hence they become potentially crucial targets for advance drug uncovering projects as we can design new inhibitors which can act more efficiently with them and may be an excellent inhibitor than inhibitors named as 0LM, 0LL, 0LK and 0LJ respectively.
Acknowledgement
we acknowledge the Department of Science and Technology (DST), New Delhi for the computational facilities in the form of FIST scheme.
References
- Rife, B.; Salemi, M.; On the early dynamics and spread of HIV-1. Trends Micro biol., 2015, 23(1), 3-4.
CrossRef - Fauci, A.S.; Marston, H.D.; Ending the HIV–AIDS Pandemic—Follow the Science. New Engl J Med., 2015, 373(23), 2197-933.
CrossRef - BarréSinoussi, F.; Ross, A.L.; Delfraissy, J.F.; Past, present and future: 30 years of HIV research. Nat Rev Microbio., 2013, 11(12), 877-83.
CrossRef - Dufloo, J.; Bruel, T.; Schwartz, O.; HIV-1 cell to-to-cell transmission and broadly neutralizing antibodies Retrovirology., 2018, 15(1), 51-60.
CrossRef - Real, F,; Sennepin, A.; Ganor, Y.; Schmitt, A.; Bomsel, M.; Live Imaging of HIV-1 Transfer across T CellVirological Synapse to Epithelial Cells that Promotes Stromal Macrophage Infection., 2018, 23(6), 1794-1805.
- Craig, W.; John, C.; Robert, W.; HIV: Cell Binding and Entry. Cold Spring Harb Perspect Med., 2012, 2(8), a006866.
- Lu, Q.; Zhang, X.; Almaula, N.; The gene for nucleoside diphosphate kinase functions as a mutator gene in Escherichia coli. J. Mol.Biol., 1995 254(3), 337-41
CrossRef - Blacklow, S.C.; Lu, M.; Kim, P.S.; A trimeric subdomain of the simian immunodeficiency virus envelope glycoprotein. Biochemistry., 1995, 34(46), 14955-14962
CrossRef - Blacklow, S.C.; Lu, M.; Kim, P.S.; A trimeric subdomain of the simian immunodeficiency virus envelope glycoprotein. Biochemistry., 1995, 34(46), 14955-14962.
CrossRef - Dalgleish, A.G.; Beverley, P.C.L.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; Weiss, R.A.; The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature.,1984 312(5996), 763-767.
CrossRef - Lasky, L. A.; Cell 1987 50(3), 975−985.
CrossRef - Blacklow, S.C.; Lu, M.; Kim, P.S.; A trimeric subdomain of the simian immunodeficiency virus envelope glycoprotein. Biochemistry., 1995, 34(46), 14955-14962
CrossRef - Kwong, P. D.; Wyatt, R.; Majeed, S.; Robinson, J.; Sweet, R. W.; Sodroski, J.; Hendrickson, W.; A. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure (London)., 2000, 8(12), 1329−1339.
CrossRef - Kwong, P. D.; Wyatt, R.; Robinson, J.; Sweet, R. W.; Sodroski, J.; Hendrickson, W. A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature., 1998, 393(6686), 648−659.
CrossRef - Zhao, Q.; Ma, L.; Jiang, S.; Lu, H.; Liu, S.; He, Y.; Strick, N.; Neamati, N.; Debnath, A.K. IdentificationofN-phenyl-′-(2,2,6,6-tetramethyl-piperidine-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology., 2005, 339(2), 213−225.
CrossRef - Lalonde, J.M.; , Kwon, Y.D.. Structure-Based Design, Synthesis, and characterization of Dual Hotspot Small-Molecule HIV-1 Entry Inhibitors. J.Med.Chem., 2012, 55(9), 4382-4396
CrossRef - Case, D.A.; Cheatham, T.E.; Daren, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J Comput Chem., 2005, 26(16), 1668–1688
CrossRef - Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem., 1993, 97(40), 10269– 10280
CrossRef - Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J Am Chem Soc., 1993, 115(21), 9620–9631
CrossRef - Jorgenson, W.L.; Chandrashekhar, J.; Madura, J.D.; Imprey, R.W.; Klein, M. Comparison of simple potential functions for simulating liquid water. J Chem Phys., 1983, 79(2), 926–935
CrossRef - Izaguirre, J.A.; Catarello, D.P.; Wozanaik, J.M.; Skeel, R.D. (2001) Langevin stabilization of molecular dynamics. J Chem Phys., 2001, 114(5), 2090–2098
CrossRef - Berendsen, H.J.C.; Postama, J.P.M.; van-Gunsteren, W.F.; DiNola, A.; Haak, J.R. ( Molecular dynamics with coupling to an external bath. J Chem Phys., 1984, 81(8), 3684–3690
CrossRef - Ryckaert, J.P.; Cicotti, G.; Barendsen, H.J.C. Numerical integration of the Cartesian equation of motion of a system with constraints: Molecular dynamics of n-alkanes. J Comput Phys., 1997, 23(3), 327–341
CrossRef - Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems, J Chem Phys., 1993, 98(12), 10089–10092
CrossRef - Humphrey, W.; Dalke, A.; Schulten, K. VMD – virtual molecular dynamics. J Mol Graph Model., 1996, 14(1), 33–38.
CrossRef - Pettersen, E.F.; Goddard, T.D.; Haung, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera (2004) A visualization system for exploratory research and analysis. J Comput Chem., 2004, 25(13), 1605–1612.
CrossRef - Tsui, V.; Case, D.A.; Theory and application of generalized born solvation model in macromolecular simulations. Biopolymers., 2001, 56(4), 271–291
- Gohlke, H.; Case, D.A. (2003) Converging free energy estimates: MM-PB (GB) SA studies on the protein-protein complex Ras-Raf. J Comput Chem, 2003, 25(2), 238–250
CrossRef - Fogolari, F.; Brigo, A.; Molinari, H. Protocols for MM/PBSA molecular dynamics simulations of proteins. Biophys J., 2003, 85(1), 159–166
CrossRef - Dubey, K.D.; Chaubey, A.K.; Ojha, R.P. Role of pH on dimeric interactions for DENV envelope protein: An insight from molecular dynamics study. Biochim Biophys Acta., 2011, 1814(12), 1796-1801
CrossRef - Dubey, K.D.; Chaubey, A.K.; Ojha, R.P. Stability of trimeric DENV envelope protein at low and neutral pH: an insight from MD study. Biochim Biophys Acta., 2013, 1834(1), 53-64
CrossRef - Dubey, K.D.; Tiwari, G.; Ojha, R.P. Targeting domain-III hinging of dengue envelope (DENV-2) protein by MD simulations, docking and free energy calculations. J Mol Model., 2017, 23(4), 102.
CrossRef - Pandey, V.; Tiwari, G.; Ojha, R. P. International Journal of Pure and Applied Physics., 2017, 13(3), 435-450.
- Mall, V. S.; Tiwari, R. K.; AIP Conference Proceedings., 2018, 1953, 140118 https://doi.org/10.1063/1.5033293.17.org/10.1063/1.5033293.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


