Synthesis, Characterization and Physicochemical Analysis of some Mannofuranoside Derivatives with Potent Antimicrobial Activity

1Department of Chemistry, Kursi Road, Integral University, Lucknow-226026, India.
2Department of Biosciences, Kursi Road, Integral University, Lucknow-226026, India.
3Department of Pharmacology and Toxicology, College of Pharmacy University of Hail, PO Box no. 2440, Saudi Arabia.
Corresponding Author E-mail: firojhassan42@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/330606
Download this article as:
![]()
We have synthesized protected mannofuranose as a glycosyl donor and some heterocyclic moieties as glycosyl acceptor. Coupling of glycosyl donor and acceptor by glycosylation, results in the formation of mannofuranoside derivatives. Antimicrobial potential of synthesized compounds were tested against five different human pathogenic bacteria and two fungi by using microdilution method. Interestingly, all synthesized mannofuranoside derivatives gave antimicrobial activity. Cumulatively, inhibitory concentration (IC50) against bacteria were found to be in the range of 84.28-309.43 μg/ml, while, IC50 against fungus were in the range of 0.59-3.82 mg/ml. Ampicillin and Fluconazole were used as positive control. Further, physicochemical parameters of synthesized mannofuranoside derivatives were analyzed using in-silico approach. Fortunately, none of our synthesized mannofuranoside derivatives gave violation of Lipinski’s Rule of Five. Thus, we can safely state that these synthesized mannofuranoside derivatives could be considered as potential anti-microbial drug candidates.
KEYWORDS:Glycosylation; Mannofuranoside; Antibacterial; Antifungal; Physicochemical parameter
Introduction
Microbial food contaminations have been the cause of severe public health concern over the last few decades1 hence antibacterial and antifungal agents are necessary for food preservation. Many researchers have focused their research on finding high potential antibacterial and antifungal agents2. The potential of sugars as starting compounds for highly efficient syntheses of carbohydrate conjugates are now well recognized with regard to antibacterial, antiviral, antineoplastic, antiprotozoal and antifungal activity3-9. Heterocyclic chemistry has wide array of biologically active compounds isolated from natural sources or synthesized in the laboratory, such as coumarins10-12, quinolines13,14, triazoles15 and phenolics16,17 etc. They are also if present as a part of giant molecule, imparting significant activity to these molecules.From the previous studies, it is realized that heterocyclic and aromatic rings are coupled with carbohydrate to enhance the biological activity18 like some acyl-substituted D-glucofuranose were discovered as more biologically active than D-glucofuranose.19 furthermore, when biologically active molecule is connected to another nuclei may possess excellent biological activity.20
Several glycoconjugates such as glycolipids, glycoproteins or glycopeptides & glycosylated natural products are frequently being used as antimicrobial drugs and also emerging as anti-cancer drug candidates21-28. Encouraged by these results, we have synthesized some glycosides for antibacterial and antifungal evaluation. During the synthesis of glycosides, protection of a particular functional group of monosaccharides is not only necessary for the modification of the remaining functional groups but also useful during the synthesis of novel derivatives of great importance29,30. Glycosylation of alcohol-acceptor precursor as heterocycles, with O-(2,3,5,6-di-O-α-D-mannofuranosyl) trichloroacetimidate36, as donor precursor in the presence of catalyst trimethylsilyl trifluoromethanesulfonate (TMSOTf) to give glycosidic products31, further antimicrobial activity of synthesized compounds were evaluated using various strains of bacteria & fungi to determine Inhibitory Concentration (IC50)32-35.
Materials and Methods
All the synthesized compounds were routinely checked for their purity on silica gel 60 (E. Merck) TLC plates and their spots were visualized by charring the plates with 5% H2SO4 in MeOH. Column chromatography on Merck silica gel (100-200 mesh) was used for the purification of synthesized compounds. Combined organic extract were dehydrated using anhydrous Na2SO4 and dried solutions were evaporated under vacuum. Solvents used in column chromatography were purchased from E. Merck, sd fine chemicals and Qualigens. Reagents used in the synthesis were purchased from, Avara syntheses, Sigma Aldrich and Himedia.
Apparatus
Electronics India, model-934 used for the determination of Melting points. ES mass spectra were recorded in Merck M-8000 LCMS system or Micromass Quadro LCMS system and HR/EI mass were done on JEOL-600H at 70eV. Proton NMR and 13C-NMR spectra were recorded on Bruker Avance DPX-300 MHz or Avance DPX-200 MHz FT Bruker spectrometers, using deuteriated solvents and Tetramethyl Silane as an internal standard. The chemical shift values from downfield to upfield in both proton NMR and 13C-NMR spectra. For synthesized compounds, proton NMR data is recorded in the order; chemical shifts values in ppm (δ), multiplicities are indicated as bs (broadened singlet), s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet) and m (multiplet). Coupling constant ‘J’ reported in Hz. Perkin-Elmer’s Spectrum RX I FT-IR spectrophotometer was used for recording IR spectra using KBr disc. Elemental analysis was carried out on Carlo-Erba-1108 instrument.
Results and Discussion
The glycosyl donor was synthesized from D-mannose, led to the formation of 2,3,5,6-di-O-isopropylidene-α-D-mannofuranose (C1). The later was treated with trichloroacetonitrile to gave 2,3,5,6-di-O-isopropylidene-1-O-trichloroacetamidyl-α-D-mannofuranose (C2) in Scheme-136.
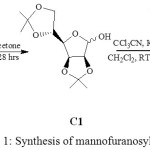 |
Scheme 1: Synthesis of mannofuranosyl donor36 |
The structure of the C1 was in accordance with its spectroscopic data such as proton NMR, isopropylidene group (>C(CH3)2) at δ 1.46-1.33 ppm.13C NMR spectrum displayed signal of methyl carbons of isopropylidene group (>C(CH3)2) at δ 25.8-27.0 ppm. The mass spectra gave a peak at 261 (M+1). FT-IR for –OH at 3431 cm-1 with 87% yield. The synthesized glycosyl donor (C2) was established by proton NMR signals at δ 8.59 ppm for –NH group,13C NMR spectrum displayed signal of methyl carbons of isopropylidene group (>C(CH3)2) δ 27.1-24.9 ppm, at δ 160.8 ppm for acetamidyl carbon. Mass spectrum gave a peak at 403 (M+1), FT-IR for –NH group was 3794 cm-1 with 66% yield.
The aglycon 7-(2-hydroxyethoxy)-4-methyl coumarin (C3) was synthesized by the reaction of 7-hydroxy-4-methyl coumarin with 2-bromoethanol and potassium carbonate in the presence of ethanol in Scheme-2.37
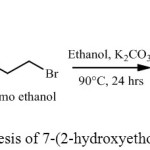 |
Scheme 2: Synthesis of 7-(2-hydroxyethoxy)-4-methyl coumarin |
The structure of C3 was established by proton NMR,13C NMR, Mass and FT-IR. Proton NMR spectra gave a triplet at δ 3.69 ppm, (-CH2OH), and δ 4.33 ppm (-CH2OAr). 13C NMR spectrum displayed a signal at δ 61.3 ppm (-CH2OH), at δ 69.9 ppm (-CH2OAr) and at δ 161.9 ppm for carbonyl carbon, Mass spectrum gave a peak at 221 (M+1), FT-IR gave frequency 3452 cm-1 (-OH), 1720cm-1 (>C=O) for compound C3 with 58 % yield.
Glycosylation of C3 as an alcohol-acceptor, with 2,3,5,6-di-O-isopropylidene-1-O-trichloroacetamidyl-α-D-mannofuranose, C2 as donor in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) and 4Aº molecular sieve in anhydrous dichloromethane gave 7-O-ethoxy-α-(2,3,5,6-di-O-isopropylidene-mannofuranosyl)-4-methyl-coumarin (C4) (Scheme-3)as colorless crystalline solid. Chemical structure of the compound C4 was confirmed by proton NMR, 13C NMR, Mass, FT-IR and elemental analysis. Proton NMR spectra revealed a signals of isopropylidene proton (>C(CH3)2) singlet at δ 1.49-1.32 ppm, singlet at δ 1.97 ppm (-CH3), triplet at δ 3.69 ppm (-CH2CH2OAr) and also a triplet at δ 4.33 ppm (-CH2 CH2 OAr). 13C NMR spectrum displayed signal of methyl carbons of isopropylidene group (>C(CH3)2) at δ 26.8-26.0 ppm, at δ 18.9 ppm (-CH3), at δ 61.3 ppm (-CH2CH2OAr), at δ 69.8 ppm (-CH2CH2OAr) and at δ 164.1 ppm for carbonyl carbon, Mass spectrum gave a peak at 463 (M+1), FT-IR gave frequency at 1720 cm-1 (>C=O), 3280, 2988cm-1 (-CH3 and -CH2 stretching), while frequency for -OH was absent. The proton NMR signals of –OH and –NH was absent in C4, formed with 59 % yield.
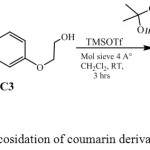 |
Scheme 3: Catalysed glycosidation of coumarin derivatives with glycosyl donor |
Synthesized glycosyl donor of D-mannose C2 was reacted with Glycosyl acceptor vanillin in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf), 4 Aº molecular sieve in anhydrous dichloromethane to formed 3-O-methoxy-4′-O-α-(2,3,5,6-di-O-isopropylidene- mannofuranosyl) benzoic acid (C5) Chemical structure of the compound C5 was confirmed by proton NMR, and FT-IR. Proton NMRspectra gave signals of isopropylidene proton (>C(CH3)2) singlet at δ 1.56-1.32 ppm, singlet at δ 3.92 ppm (-OCH3) and also a singlet at δ 11.74 ppm (-COOH). 13C NMR spectrum displayed signal of methyl carbons of isopropylidene group (>C(CH3)2) at δ 26.2-23.8 ppm, at δ 56.2 ppm (-OCH3) and at δ 169.5 ppm for carbonyl carbon, Mass spectrum gave a peak at 411 (M+1), FT-IR gave frequency at 1690 cm-1 (>C=O), 3298, 2925 and 2870 cm-1 (-CH3 and –CH2 stretching), while frequency for -OH was absent. The proton NMR signals of -OH and –NH was absent in C5. It was formed with 45 % yield.
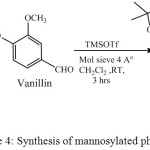 |
Scheme 4: Synthesis of mannosylated phenolics |
Synthesized glycosyl donor C2 was coupled with 8-Hydroxy quinoline, 1-Hydroxy benzotriazole and 2-(Hydroxymethyl) furan in the presence of catalytic amount of trimethylsilyl trifluoromethanesulfonate (TMSOTf), 4Aº molecular sieve in anhydrous dichloromethane at room temperature to gave 8-O-α-(2,3,5,6-di-O-isopropylidene-mannofuranosyl)-quinoline (C6), 1-O-α-(2,3,5,6-di-O-isopropylidene-mannofuranosyl)-1H-benzo-1,2,3-triazole (C7) and 2-O-methyl-α-(2,3,5,6-di-O-isopropylidene-mannofuranosyl) furan (C8) with 56, 67, 69% yield respectively. Structure of these compounds was established by proton NMR, 13C NMR, Mass and FT-IR.
Proton NMR spectra of C6 gave signals of isopropylidene proton (>C(CH3)2) singlet at δ 1.36-1.25 ppm, doublet at δ 8.73 ppm (H-1), triplet at δ 7.52 ppm (H-2), doublet at δ 7.58 ppm (H-3) and multiplet at δ 7.41-7.16 ppm.13C NMR spectrum displayed signal of methyl carbons of isopropylidene group (>C(CH3)2) at δ 27.0-24.7 ppm, carbon of aromatic ring was observed at δ 163.9, 152.3, 148.0, 138.4, 136.3, 128.7, 127.9, 122.0, 118.1 ppm, Mass spectrum gave a peak at 388 (M+1), FT-IR gave frequency at 2988 and 2881 cm-1 (-CH3 and –CH2 stretching), while frequency for -OH was absent.
Proton NMR spectra of C7 gave signals of isopropylidene proton (>C(CH3)2) at δ 1.47-1.31 ppm and multiplet at δ 7.69-7.38 ppm (Ar-H). 13C NMR spectrum displayed signal of methyl carbons of isopropylidene group (>C(CH3)2) at δ 26.0-24.7 ppm, carbons of aromatic ring was observed at δ 144.8, 144.7, 135.1, 130.8, 129.8, 129.6, 128.8, ppm, Mass spectrum gave a peak at 378 (M+1), FT-IR gave frequency at 3039 and 2917 cm-1 (-CH3 and –CH2 stretching), while frequency for -OH was absent.
Proton NMR spectra of C8 gave signals of isopropylidene proton (>C(CH3)2) singlet at δ 1.42-1.329 ppm, singlet at δ 3.92 ppm (H-1), doublet at δ 6.24 ppm (H-2), triplet at δ 6.27ppm (H-3), doublet at δ 6.98 ppm (H-4). 13C NMR spectrum displayed signal of methyl carbons of isopropylidene group (>C(CH3)2) at δ 26.0-24.7 ppm, carbons of furan ring was observed at δ 152.8, 143.8, 113.7, 112.7 ppm, Mass spectrum gave a peak at 341 (M+1), FT-IR gave frequency at 3282 and 2974 cm-1 (-CH3 and –CH2 stretching), while frequency for -OH was absent. The proton NMRsignals of –OH and –NH was absent in C6, C7 and C8.
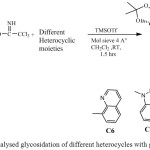 |
Scheme 5: Catalysed glycosidation of different heterocycles with glycosyl donor |
Antibacterial Activity
Determination of Inhibitory Concentration (IC50)
The disk diffusion method38 was used for the preliminary antibacterial evaluation of compounds C1, C2, C4-C8. The inhibitory concentrations (IC50) of mannofuranoside derivatives showing inhibition in the preliminary tests, were determined by the microtitre plate technique using micro dilution method (Table-1)39 Briefly, Staphylococcus aureus (ATCC 6538), Bacillus pumilis (MTCC 160), Bacillus subtilis (MTCC 441), Bacillus cerius (MTCC1305), Escherichia coli (ATCC 25923), obtained from NCIM NCL Pune, India were grown to mid-logarithmic phase and harvested by centrifugation, washed with 10 mM sodium phosphate buffer (SPB) at pH 7.4, and diluted to 2 x 105 colony forming units (CFU) ⁄ ml in SPB containing 0.03% Nutrient broth (NB). Mannofuranoside derivatives were serially diluted in 250 µL of nutrient broth (NB) medium in 96-well microtitre plates to achieve the desired concentrations (25-400µg ⁄ ml) with bacterial inoculum (5 x 104 CFU per well). After incubation at 37ºC overnight, the IC50 was taken of the synthesized compound that provides a 50% reduction in growth of bacterial strains compared to the growth in the control well.
Antifungal Activity
Determination of Inhibitory Concentration (IC50)
The utilization of microplate bioassays, or broth microdilution tests, to measure the biological action of synthetic compounds against fungal pathogens has expanded in recent years. This method has been recognized as the most encouraging invitro bioassay for evaluating antifungal activity 40,41. The antifungal strains A. flavus (MTCC 277) and T. viridens (MTCC 167) for the test were obtained from NCIM, NCL Pune, India. Mannofuranoside derivatives were serially diluted in 250 µL of SD medium in 96-well microtitre plates to achieve the desired concentrations (0.5-5 mg/ml) with fungal inoculum (5 x 102 CFU per well). The invitro antifungal activities of the test compounds under investigation were also done by Poisons Food technique42 and the technique was used with some modification by43 using 0.5-5 mg of compounds per ml of Sabouraud Dextrose (SD) medium. The diameter of radial growth of the test fungi was measured after 2 to 3 days of incubation at 27±1ºC and expressed as percent mycelia growth inhibition following the formula given below:
I = C-T/CX100
Where, I = Percentage of inhibition.
C = Optical density of the fungal colony at 595 nm in control (DMSO)
T = Optical density of the fungal colony in treatment.
Calculation of Physicochemical Properties
To check the physicochemical properties of synthesized mannofuranoside derivatives, Molinspiration property count instrument (http://www.molinspiration.com/cgi-bin/properties) was utilized. Different parameters as topological polar surface area (TPSA), molecular weight, miLogP, the quantity of hydrogen bond donors, the number of hydrogen bond acceptors, number of rotatable bonds, and violations of Lipinski’s rule of five44 were estimated using Molinspiration tool [Table-2]. Moreover, Absorption % was ascertained by the strategy for Zhao et al. (2002) utilizing the recipe
% of Absorption = 109 ‒ [0.345 × TPSA]
Physicochemical Properties Prediction
Earlier, lead drug compounds fails at different clinical trials stages as a result of inappropriate pharmacokinetic profiling. But, now- a- days to cut down these failures, physicochemical profiling of new drug candidates has become an important part of drug discovery. These analyses involve assessment of various molecular descriptors that would help in prediction of drug-likeliness. There are various in- silico tools available to calculate the molecular properties of a compound to assess its pharmacokinetic profile. In fact, these in-silico tools have helped a lot to overcome the issues related to the failure of clinical trials45,46.
Molinspiration is a tool which was used in the present study to screen the pharmacokinetic property of mannofuranoside derivatives i.e., C1, C2, C4-C8. According to the Lipinski’s Rule of Five (Lipinski et al., 2001) studied mannofuranoside derivatives posses’ drug like property [Table-2]. Lipinski’s Rule of Five is based on the observation that the majority of oral drugs have a molecular weight ≥500, Log P value ≥5, hydrogen bond donor ≥5, hydrogen bond acceptor ≥10 and rotatable bonds ≥10. Any compound which violates more than one of these rules might have problem. Interestingly, none of our synthesized mannofuranoside derivatives gave violation of Lipinski’s Rule of Five. Thus, we can safely state that these compounds could be considered as potential drug candidates.
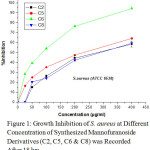 |
Figure 1: Growth Inhibition of S. aureus at Different Concentration of Synthesized Mannofuranoside Derivatives (C2, C5, C6 & C8) was Recorded After 18 hrs. |
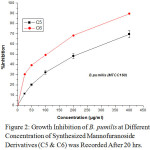 |
Figure 2: Growth Inhibition of B. pumilis at Different Concentration of Synthesized Mannofuranoside Derivatives (C5 & C6) was Recorded After 20 hrs. |
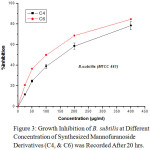 |
Figure 3: Growth Inhibition of B. subtilis at Different Concentration of Synthesized Mannofuranoside Derivatives (C4, & C6) was Recorded After 20 hrs. |
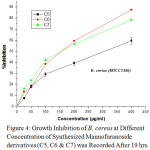 |
Figure 4: Growth Inhibition of B. cereus at Different Concentration of Synthesized Mannofuranoside derivatives (C5, C6 & C7) was Recorded After 19 hrs. |
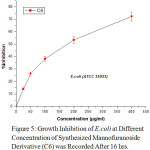 |
Figure 5: Growth Inhibition of E.coli at Different Concentration of Synthesized Mannofuranoside Derivative (C6) was Recorded After 16 hrs. |
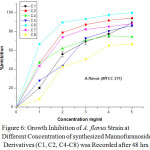 |
Figure 6: Growth Inhibition of A. flavus Strain at Different Concentration of synthesized Mannofuranoside Derivatives (C1, C2, C4-C8) was Recorded after 48 hrs. |
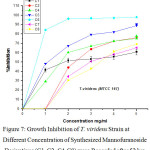 |
Figure 7: Growth Inhibition of T. viridens Strain at Different Concentration of Synthesized Mannofuranoside Derivatives (C1, C2, C4-C8) were Recorded after 52 hrs. |
The growth inhibition of different strains of bacteria (S. aureus (ATCC 6538), B. pumilis (MTCC 160), B. subtilis (MTCC 441), B. cerius (MTCC1305), E. coli (ATCC 25923) and fungi A. flavus (MTCC 277), T. viridens (MTCC 167) obtained from NCIM, National chemical laboratories, Pune) at different concentration were presented in Fig. 1-7.
Table 1: Antibacterial (IC50 µg/ml) and Antifungal activity (*IC50 mg/ml) of Compounds C1, C2, C4-C8:
|
Compounds |
S. aureus |
B. pumilis |
B. Subtilis |
B. cerius |
E.coli |
A. flavus |
T. viridens |
|
C1 |
NA |
NA |
NA |
NA |
NA |
1.84(±0.04) |
1.86(±0.02) |
|
C2 |
267.67(±0.17) |
NA |
NA |
NA |
NA |
1.09(±0.01) |
2.30(±0.05) |
|
C4 |
NA |
NA |
157.61(±0.32) |
NA |
NA |
1.18(±0.05) |
1.66(±0.03) |
|
C5 |
234.62(±0.16) |
221.65(±0.25) |
NA |
309.43(±0.19) |
NA |
2.21(±0.03) |
1.08(±0.01) |
|
C6 |
84.28(±0.11) |
101.12(±0.13) |
102.68(±0.16) |
154.78(±0.21) |
178.96(±0.31) |
0.75(±0.02) |
0.59(±0.01) |
|
C7 |
NA |
NA |
NA |
156.67(±0.34) |
NA |
1.23(±0.05) |
2.98(±0.04) |
|
C8 |
294.15(±0.31) |
NA |
NA |
NA |
NA |
2.91(±0.02) |
3.82(±0.03) |
|
Ampicillin |
4.14(±0.7) |
14.15(±0.06) |
11.11(±0.09) |
8.51(±0.42) |
28.03(±0.10) |
– |
– |
|
Fluconazole |
– |
– |
– |
– |
– |
0.10(±0.01) |
0.08(±0.01) |
NA, not active, (±) shows SD
Table 2: Physicochemical Parameters of Compounds C1, C2, C4-C8 and standard drugs
|
Compounds |
Physiochemical parameters |
|||||||
|
% Absa |
TPSAb |
MWc |
miLogPd |
HBDe |
HBAf |
RBg |
Lip. vioi |
|
|
Rule |
– |
– |
<500 |
≤5 |
<5 |
<10 |
≤10 |
≤1 |
|
C1 |
86.09 |
66.40 |
260.29 |
0.70 |
1 |
6 |
1 |
0 |
|
C2 |
81.65 |
79.26 |
404.67 |
2.85 |
1 |
7 |
4 |
0 |
|
C4 |
76.27 |
94.85 |
462.50 |
3.30 |
0 |
9 |
6 |
0 |
|
C5 |
73.83 |
101.94 |
410.42 |
2.52 |
1 |
9 |
5 |
0 |
|
C6 |
85.43 |
68.30 |
387.43 |
2.97 |
0 |
7 |
3 |
0 |
|
C7 |
79.28 |
86.12 |
377.40 |
2.34 |
0 |
9 |
3 |
0 |
|
C8 |
85.35 |
68.54 |
340.37 |
2.17 |
0 |
7 |
4 |
0 |
|
Ampicillin |
70.10 |
112.73 |
349.41 |
-0.87 |
4 |
7 |
4 |
0 |
|
Fluconazole |
80.82 |
81.66 |
306.88 |
-0.12 |
1 |
7 |
5 |
0 |
% Absa = % of Absorption= 109 ‒ [0.345 × Topologicl Polar Surface Area]
TPSAb = Topological Polar Surface Area (Å)2
MWc = Molecular Weight
miLogPd = Logarithm of compound partition coefficient between n-octanol and water
HBDe = Hydrogen Bond Donors
HBAf = Hydrogen Bond Acceptors
RBg = Rotatable bond
Lip. vioi = Lipinski’s Violation
Experimental
Synthesis of 2,3,5,6-di-O-isopropylidene-α-D-mannofuranose (C1)36
Yield 87%, Rf 0.74, mp = 124-126⁰C; Proton NMR(300 MHz, CDCl3): δ 4.83-4.79 (m, 1H); δ 4.46-4.42 (m, 1H); δ4.61-4.63 (d, J = 5.6 Hz, 1H,); 4.18-4.21 (m, 1H); δ 4.05-4.17 (dd, J = 2.1 Hz, 1.2 Hz, 2H); δ 3.1 (s, 1H); δ 1.47, δ1.46, δ1.38 & δ 1.33 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 113.4, 112.7, 109.4, 109.2, 101.2, 101.0, 97.2, 85.6, 80.1, 79.7, 79.4, 79.1, 78.6, 76.8, 76.2, 73.4, 73.0, 70.1, 67.1, 27.0, 26.8, 25.9, 25.8; MS(m/z) = 261 (M+1); IR (KBr): (υ cm-1): 3431, 2982, 2875; Anal. Calcd for C12H20O6: Calculated C, 55.37; H, 7.74 Found C, 55.29; H, 7.71
Synthesis of 2, 3, 5, 6-di-O-isopropylidene-1-O-trichloroacetamidyl-α-D-mannofuranose (C2)36
Yield 66%, Rf 0.92, mp = 103-105⁰C; Proton NMR(300 MHz, CDCl3): δ 8.59 (s, 1H, -NH); δ 6.26 (s, 1H); δ 4.94 (m, 2H); δ 4.42 (m, 1H); 4.04 (m, 3H); δ 4.05-4.17 (dd, J = 2.1 Hz, 1.2 Hz, 2H); δ 1.59, δ 1.50 , δ 1.45 & δ 1.33 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ: 160.8, 113.6, 112.7, 109.6, 109.2, 104.9, 101.4, 91.1, 85.6, 84.9, 83.0, 80.5, 79.8, 79.4, 79.1, 76.8, 73.4, 72.8, 67.2, 66.8, 27.1, 26.1, 25.3, 24.9; MS(m/z) = 404 (M+1); IR (KBr): (υ cm-1): 3794, 3334, 2988 Anal. Calcd for C14H20O6NCl3: Calculated C, 40.68; H, 4.98; N, 3.46; Found C, 41.08, H, 4.81, N 3.38.
Synthesis of 7-(2-hydroxyethoxy)-4-methyl coumarin (C3)
A mixture 7-hydroxyl-4-methyl coumarin (5.0 gm, 22.72 mmol), 2-bromoethanol (5.0 gm, 40.65 mmol) and potassium carbonate (3.0 gm, 21.89 mmol) are taken in flat bottom flask (25 ml) and refluxed for 24 hrs in ethanol (100 ml). After cooling treated with ether and water (1:1) then separated. The aqueous layer further washed with ether (2 x 20 ml), both organic layers combined; add sodium sulphate to dry and solvent evaporate under reduced pressure obtained residue was directly allow to column chromatography using hexane/ethyl acetate (4/6) to give the desired compound C3 with interesting yield for crystallization ether was added and 7-(2-hydroxyethoxy)-4-methyl coumarin obtained in the form of white crystalline solid.
Yield 58%, mp = 128-130⁰C; Proton NMR(300 MHz, CDCl3): δ 7.67 (d, J = 1.5 Hz, 1H); δ 6.83(d, J = 2.3 Hz, 1H); δ 6.93 & δ 6.91 (dd, 1.6 & 1.2 Hz, 1H); δ 6.57 (s, 1H); δ 1.97 (s, 3H); δ 2.6 (s, 1H,); δ 3.69 (t, 2H); δ 4.33 (t, 2H); 13C NMR (75 MHz, CDCl3) δ: 161.9, 161.6, 155.3, 152.8, 125.8, 114.1, 113.3, 112.7, 112.3, 111.6, 103.5, 101.8, 69.9, 61.3, 19.3; MS (m/z) = 221 (M+1); IR (KBr) (υ cm-1): 3452; 3021, 2982, 1720, Anal. Calcd for C11H8O4: Calculated C, 64.07; H, 4.89; Found C, 64.01, H, 4.74.
Synthesis of 7-O-ethoxy-α–(2,3,5,6-di-O-isopropylidene-mannofuranosyl)-4-methyl-coumarin (C4)
To synthesized C4, the mixture of C2 (1.21 gm, 3.0 mmol) and C3 (0.61 gm, 3.0 mmol) was stirred in dry dichloromethane CH2Cl2 (15 ml) in the presence of trimethylsilyl trifluoromethanesulfonate TMSOTf (60 µl, 0.30 mmol) and molecular sieve (1.0 gm, 4Aº) for 1.5 h at room temperature and then solvent evaporated under reduced pressure obtained residue was directly allow to column chromatography using hexane/ethyl acetate (9/1) to give the desired compound C4 as white crystalline solid with interesting yield.
Yield 49%, Rf 0.62, mp = 116-118⁰C; Proton NMR(300 MHz CDCl3): δ 7.67 (d, J = 1.5 Hz, 1H); δ 6.83 (d, J = 2.3 Hz, 1H); δ 6.91-6.89 (dd, 1.6 & 1.5 Hz, 1H); δ 6.57 (s, 1H); δ 5.62 (s, 1H), δ 4.87 (m, 1H); δ 4.91 (m, 1H) δ 4.42-4.39 (m, 1H); δ 4.62-4.60 (d, J = 7.5 Hz, 1H); δ 4.16-4 .18 (dd, J = 2.0 Hz, 1.5 Hz, 2H); δ 1.97 (s, 3H); δ 3.69 (t, 2H); δ 4.33 (t, 2H); δ 1.49, δ 1.43, δ 1.35 & δ 1.32 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 164.1, 161.9, 160.2, 126.1, 125.9, 113.3, 112.8, 112.2, 111.5, 103.5, 101.7, 91.9, 69.8, 61.3, 29.8, 27.0, 26.0, 26.3, 24.7, 18.9; MS(m/z) = 463 (M+1); IR (KBr) (υ cm-1): 3282, 3021, 2974, 1720 ;Anal. Calcd for C24H30O9: Calculated C, 62.33; H, 6.54; Found C, 64.46, H, 6.62.
Synthesis of 3-O-methoxy-4′-O-α-(2,3,5,6-di-O-isopropylidene- mannofuranosyl) benzoic acid (C5)
To synthesized C5, the mixture of C2 (1.21 gm, 3.0 mmol) and vanillin (0.50 gm, 3.0 mmol) was stirred in dry dichloromethane CH2Cl2 (15 ml) in the presence of trimethylsilyl trifluoromethanesulfonate TMSOTf (60 µl, 0.30 mmol) and molecular sieve (1.0 gm, 4Aº) for 2.5 h at room temperature and then solvent evaporated under reduced pressure obtained residue was directly allow to column chromatography using hexane/ethyl acetate (9/1) to give the desired compound C5 as light yellow crystalline solid with sufficient yield.
Yield 45%, Rf 0.42, mp = 97-99⁰C; Proton NMR(300 MHz CDCl3): δ 11.74 (s, 1H); δ 7.81 (d, J = 1.5 Hz, 1H); δ 7.41-7.38 (dd, J = 1.5 Hz & 1.2 Hz); δ 7.16 (d, J = 1.5 Hz, 1H); δ 5.69 (s, 1H), δ 4.93 (m, 1H); δ 4.89 (m, 1H); δ 4.41-4.38 (m, 1H); δ 4.63-4.61 (d, J = 7.0 Hz, 1H); δ 4.13-4 .19 (dd, J = 2.2 Hz, 1.2 Hz, 2H); δ 3.92 (s, 3H); δ 1.56, δ 1.53, δ 1.36 & δ 1.32 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 169.5, 151.1, 145.8, 144.7, 126.7, 124.3, 124.2, 123.8, 116.4, 115.1, 111.4, 103.6, 101.2, 85.5, 71.2, 56.2, 26.2, 25.2, 25.0, 23.8; MS(m/z) = 411 (M+1); IR (KBr) (υ cm-1): 3298, 2925, 2870, 1690;Anal. Calcd for C20H26O9: Calculated C, 58.53; H, 6.39; Found C, 58.62, H, 6.35.
Synthesis of 8-O-α–(2,3,5,6-di-O-isopropylidene-mannofuranosyl)-quinoline (C6)
To synthesized C6, the mixture of C2 (1.21 gm, 3.0 mmol) and 8-Hydroxy quinoline (0.87 gm, 6.0 mmol) was stirred in dry dichloromethane CH2Cl2 (15 ml) in the presence of trimethylsilyl trifluoromethanesulfonate TMSOTf (60 µl, 0.30 mmol) and molecular sieve (1.0 gm, 4Aº) for 1.5 h at room temperature and then solvent evaporated under reduced pressure obtained residue was directly allow to column chromatography using hexane/ethyl acetate (9/1) to give the desired compound C6 as white crystalline solid with sufficient yield.
Yield 44%, Rf 0.65, mp = 110-113⁰C; Proton NMR(300 MHz CDCl3): δ 8.73 (d, J = 7.5 Hz, 1H); δ 7.52 (t, 1.5 Hz, 1H); δ 7.40 (d, J = 1.2 Hz, 1H); δ 7.16 (d, J = 1.5 Hz, 1H); δ 7.41 (t, 1.2 Hz, 1H); δ 7.24 (d, J = 1.5 Hz, 1H); δ 5.71 (s, 1H), δ 4.98 (m, 1H); δ 4.91 (m, 1H) δ 4.42-4.39 (m, 1H); δ 4.62-4.60 (d, J = 7.5 Hz, 1H); δ 4.16-4.18 (dd, J = 2.0 Hz & 1.5 Hz, 2H); δ 1.36, δ 1.34, δ 1.27 & δ 1.25 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 163.9, 152.3, 148.0, 138.4, 136.3, 128.7, 127.9, 122.0, 118.1, 113.0, 110.2, 109.5, 109.2, 101.7, 101.5, 92.0, 85.6, 85.2, 81.1, 80.5, 79.8, 73.4, 73.1, 67.1, 66.7, 29.8, 27.0, 26.0, 25.3, 24.7; MS(m/z) = 388 (M+1); IR (KBr) (υ cm-1): 3382, 2988, 2881; Anal. Calcd for C21H25O6N: Calculated C, 65.10; H, 6.50, N, 3.62; Found C, 65.15, H, 6.59, N, 3.71.
Synthesis of 1-O-α–(2,3,5,6-di-O-isopropylidene-mannofuranosyl)-1H-benzo-1,2,3-triazole (C7)
To synthesized C7, the mixture of C2 (1.21 gm, 3.0 mmol) and 1-Hydroxy benzotriazole (0.86 gm, 6.4 mmol) was stirred in dry dichloromethane CH2Cl2 (15 ml) in the presence of trimethylsilyl trifluoromethanesulfonate TMSOTf (60 µl, 0.30 mmol) and molecular sieve (1.0 gm, 4Aº) for 1.5 h at room temperature and then solvent evaporated under reduced pressure obtained residue was directly allow to column chromatography using hexane/ethyl acetate (9/1) to give the desired compound C7 as colourless oil with sufficient yield.
Yield 48%, Rf 0.65, mp = 118-120⁰C; Proton NMR(300 MHz CDCl3):δ 7.69 (m, 2H); δ 7.38-7.56 (m, 2 H, Ar-H); δ 5.81 (s, 1H), δ 4.94 (m, 1H); δ 4.92 (m, 1H) δ 4.42-4.58 (m, 1H); δ 4.81-4.83 (d, J = 7.0 Hz, 1H); δ 4.04-4.12 (dd, J = 2.0 Hz & 1.5 Hz, 2H); δ 1.47, δ 1.40, δ 1.35 & δ 1.31 (s, 3H); 13C NMR (75 MHz, CDCl3) δ: 144.8, 144.7, 135.1, 130.8, 129.8, 129.6, 128.8, 128.1, 114.3, 114.2, 113.8, 113.2, 96.8, 96.7, 79.5, 63.2, 27.0, 26.0, 25.3, 24.8, 24.72; MS(m/z) = 378 (M+1); IR (KBr) (υ cm-1): 3039, 2917;Anal. Calcd for C18H23O6N3: Calculated C, 57.29; H, 6.14, N, 11.13; Found C, 57.23, H, 6.04, N, 11.28.
Synthesis of 2-O-methyl-α–(2,3,5,6-di-O-isopropylidene-mannofuranosyl) furan (C8)
To synthesized C8, the mixture of C2 (1.21 gm, 3.0 mmol) and 2-(Hydroxymethyl) furan (0.61 ml, 6.0 mmol) was stirred in dry dichloromethane CH2Cl2 (15 ml) in the presence of trimethylsilyl trifluoromethanesulfonate TMSOTf (60 µl, 0.30 mmol) and molecular sieve (1.0 gm, 4Aº) for 2.5 h at room temperature and then solvent evaporated under reduced pressure obtained residue was directly allow to column chromatography using hexane/ethyl acetate (9/1) to give the desired compound C8 as crystalline solid.
Yield 38%, Rf 0.59, mp = 112-114⁰C; Proton NMR(300 MHz CDCl3): δ 3.92 (s, 1H); δ 6.24 (d, d = 7.5 Hz, 1H); δ 6.27 (t, 1H); δ 6.98 (d, 1.2 Hz, 1H); δ 5.53 (s, 1H), δ 4.83 (m, 1H); δ 4.979 (m, 1H) δ 4.38-4.47 (m, 1H); δ 4.74-4.79 (d, J = 7.0 Hz, 1H); δ 4.03-4.11 (dd, J = 2.0 Hz & 1.5 Hz, 2H); δ 1.42, δ 1.35, δ 1.31 & δ 1.29; 13C NMR (75 MHz, CDCl3) δ: 152.8, 143.8, 113.7, 112.7, 109.2, 107.9, 101.2, 85.6, 80.1, 79.7, 73.0, 64.1, 26.0, 25.8, 24.9, 24.8; MS(m/z) = 341 (M+1); IR (KBr) (υ cm-1): 3282, 2974;Anal. Calcd for C17H24O7: Calculated C, 59.99; H, 7.11, N; Found C, 59.90, H, 7.16.
Structure Activity Relationship (SAR)
The synthesized compounds selected for the antimicrobial activity had the same parental skeleton. The most important things for the activity were due to substituent’s which attached to parent skeleton. The mannofuranoside derivative, 8-O-α-(2,3,5,6-di-O-isopropylidene-mannofuranosyl)-quinoline (C6) have IC50 84.28 µg/ml against S. aureus due to the presence of quinoline as heterocyclic substituent. Secondary nitrogen of the quinoline having a nucleophilic centre has been found essential for activity. The mannofuranoside derivative, Synthesis of 3-O-methoxy-4′-O-α-(2,3,5,6-di-O-isopropylidene- mannofuranosyl) benzoic acid (C5) have maximum IC50 309.43 µg/ml against B. cerius because they have ethoxy group as substituent. ethoxy group destabilize the ring and reduced activity. The mannofuranoside derivative, 8-O-α-(2,3,5,6-di-O-isopropylidene-mannofuranosyl)-quinoline (C6) have IC50 0.59 mg/ml against T. viridens due to the presence of quinoline as heterocyclic substituent. Secondary nitrogen of the quinoline having a nucleophilic centre has been found essential for activity. The mannofuranoside derivative, 2-O-methyl-α-(2,3,5,6-di-O-isopropylidene-mannofuranosyl) furan (C8) have IC50 3.82 mg/ml against T. viridens having methyl furanyl as heterocyclic substituent. Methyl group destabilize the ring and reduced activity.
Eight mannofuranoside derivatives were synthesized and IC50 was determined for all derivatives. From the IC50 value depicted in table-1 it can be concluded that mannofuranoside with quinoline as heterocyclic substituent (C6) is more potent than coumarin analogue (C4), Phenolic (C5), triazole (C7) and methyl furanyl analogue (C8).
In this study, synthesized mannofuranoside derivatives, were found to be as strong antifungal agents. In addition, these compounds inhibited few pathologically important gram positive and gram negative bacteria also. As they gave significant antifungal potential, these compounds could be further improved to form broad spectrum antifungal agents. In addition, none of these mannofuranoside derivatives gave violation of Lipinski’s Rule of Five. Further studies would be needed to decipher the exact mechanism of anti-microbial/anti-fungal action of these derivatives. However, we can safely state that the present study has provided some potent anti-microbial drug candidates in the form of mannofuranoside derivatives.
Acknowledgement
The authors are grateful to thank the R&D wing of Integral University, Lucknow, for support and provide communication number (IU/R&D/2017-MCN00021) for the manuscript and authors are also thankful to Mr. Wajid Ali, IIT, Guwahati, Ms. Deepti Saxena, Tahseen Akhtar, Javed, CDRI, Lucknow, India for their help in spectroscopic data analysis.
References
- Wilson, C.L. Microbial Food Contamination, CRC Press, Taylor & Francis Group, LLC: Boca Raton, FL, USA. 2007.
- Shen, Y.; Sun, Y.; Sang, Z.; Sun, C.; Dai, Y.; Deng, Y. Molecules. 2012, 17, 8661-8673.
CrossRef - Nelson, D.L.; Cox, M.M. Lehninger Principle of Biochemistry, Macmillan, USA. 2004, 238- 254.
- Ritter, T.K.; Wong, C.H. Angew. Chem. Int. Ed. Engl. 2001, 40, 3508-3533.
CrossRef - Nogueira, C.M.; Parmanhan, B.R.; Farias, P.P.; Correa, A.G. Rev. Virtual Quim. 2009,1,149-159.
- Wong, C. Carbohydrate-based Drug Discovery, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. 2003
- Feng, X.U.; Ding, Q.; Yang, K.; Jing, W.G. Chinese Chem. Lett. 2006, 17, 187-190.
- Gramik, V.G.; Zhidkava, A.M.; Kiselev, S.S.; Ghohkov, R.G.; Polezhaeva, A.J; Moshkovshi, M.D. J Pharm Chem. 1978, 12, 881.
CrossRef - Senthil, S.; Gopi, R. Der Pharma Chemica. 2015, 7, 15-23.
- Solon, S.; Brandao, C.A.Ce.L.F.G. Quim. Nova. 2012, 35, 1169-1172.
CrossRef - Alves, M.J.; Ferreira, I.C.F.R.; Froufe, H.J.C.; Abreu, R.M.V.; Martins, A.; Pintado, M. Journal of Applied Microbiology. 2013, 115, 346-357.
CrossRef - Gupta, R.; Paul, S.; Gupta, A.K.; Kachroo, P.L.; Bani, S. Indian J. Chem. 1997, 36, 707–710.
- Kabir, A.K.M.S.; Matin, M.M.; Bhuiyan, M.M.R.; Rahim, M.A. Chittagong Univ. J. Sci. 2001, 25, 85-94.
- Hassan, F.; Kamal, A.; Ahmad, N.; Nasibullah, M.; Khan, M.S.; Khan, A.R. Inter. J. of Chem. 2013, 34, 1395-1402.
- Nogueira, C.M.; Parmanhan, B.R.; Farias, P.P.; Corrêa, A.G. Rev.Virtual Quim. 2009, 1, 149-159.
- Cao, H.; Hwang, J.; Chen, Xi. Carbohydrate-containing natural products in medicinal chemistry. InL Edn. 2011, 411-431.
- Cipolla, L.; La Ferla, B.; Airoldi, C.; Zona, C.; Orsato, A.; Shaikh, N.; Russo, L.; Nicotra, F. Fut. Med. Chem. 2010, 2, 587-599.
CrossRef - Cipolla, L.; Gregori, M.; So, P.W. Curr. Med. Chem. 2011, 8, 1002-1018.
CrossRef - Cipolla, L.; Peri, F. Mini-Rev. Med. Chem. 2011, 11, 39-54.
CrossRef - Lonngren, J. Pure Appl. Chem. 1989, 61, 1313-1314.
CrossRef - Horne, G.; Wilson, F.X.; Prog. Med. Chem. 2011, 50, 135-176.
CrossRef - Etayo, P.; Badorrey, R.; Villegas, M.D.; Galvez, G.A.; Víu, P.L. Synlett. 2010, 12, 1775-1778.
- Andry, C.; Wylde, R.; Laffite, C.; Privat, G.; Winternitz, F. Phytochem. 1982, 21, 1123–1127.
CrossRef - Ishi, H.; Nakamura, M.; Seo, S.; Tori, K.; Tozyo, T. Chem. Pharm. Bull. 1980, 28, 2367–2373.
CrossRef - Schmidt, R.R.; Michel, J. Angew. Chem. 1980, 92, 763-764.
CrossRef - Ayşe, N.; Duygu, A.T.I.; Hakkı, A.; Tansel, O.Y.; İsmet, D.G; İsmail, K. FABAD J. Pharm. Sci. 2008, 33, 77–86.
- Dulger, G.; Aki, C. Trop. J.l of Pharmace. Res. 2009, 8, 371-375.
- Annette, W.F.; Rinaldi, M.G.; Sutton, D.A. Infect Dis. Clin N Am. 2006, 20, 699–709.
- Kratzer, C.; Tobudic, S.; Schmoll, M.; Graninger, W.; Georgopoulos, A. Journal of Antimicrobial Chemotherapy. 2006, 58, 1058–1061.
CrossRef - Filice, M.; Guisan, J.M.; Palomo, J.M. Curr. Org. Chem. 2010, 14, 516-532.
CrossRef - Obi, M.; Morino, S.; Ichimura, K. Chem. Mat. 1999, 11, 656-664.
CrossRef - Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turch, M. Am. J. Clin. Pathol. 1966, 4, 5493-496.
- Amsterdam, D. In Lorian V. ed. Antibiotics in Laboratory Medicine, (Lippincott Williams & Wilkins Philadelphia P A). 2005, 72–78.
- Pryor, W.S.; Gibson, M.D.; Bergstrom, C.G.; Walker, P.L. BioTechniques. 2007, 42, 168-172.
CrossRef - Hadacek, F.; Greger, H.; Phytochem. Anal. 2000, 11, 137-147.
CrossRef - Grover, R.K.; Moore, J.D. Phytopathol. 1962, 52, 876-880.
- Miah, M.A.T.; Ahmed, H.U.; Sharma, N.R.; Ali, A.; Miah, S.A.; Bangladesh. J. Bot. 1990, 19, 5–10.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Adv Drug Deliv Rev. 2001, 46, 3–26.
CrossRef - Aboelmagd, A.; Ibrahim, A.I.A.; Ezzeldin, M.S.S.; Razik, M.A. ARKIVOC. 2011, 9, 337-353.
- Mamdouh, A.S.; Samy, B.S.; Kandeel, H.S. Der Pharma Chemica. 2012, 4, 1064-1073
- Mohammed, M.M.; Bhuiyan, M.M.H.; Dulal, C.D.; Manchur, M.A. Int. J. Biosci. 2013, 3, 279-287.
CrossRef - Katritzky, A.R. In Comprehensive Heterocyclic Chemistry, Elsevier Science and Technology ISBN: 0080420729. 1996, 11628.
- Govori, S.; Rapic, V.; Leci, O.; Cacic, M.; Tabakovic, I. J. Heterocycl. Chem. 1996, 33, 351-354.
CrossRef - Govori, S.; Spahiu, S.; Kalaj, V.; Leci, O.; Haziri, A.; Ibrahimi, H. Am. Jour. of Bioch. and Biotechnol. 2010, 6, 275-278
CrossRef - Azam, F, Mohamed, N.; Alhussen, F. Network: Computation in neural systems. 2015, 26, 97-115.
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Pharm. Res. 2002, 19, 1446-1457.
CrossRef







ISSN Online: 2231-5039

![]()




{kind=link}