Synthesis, Characterization and Molecular Docking of Novel Quinoline and Pyridine Derivatives
1Department of Chemistry, Al rasafi school, Ministry of Education , Anbar, Iraq.
2Department of Chemistry, College of health medical technology, Baghdad, Iraq.
3Department of Chemistry, College of Education for Women , University of Anbar, Iraq.
4Department of Studies in Physics, Manasagangotri, University of Mysore, Mysore-570006, Karnataka, India.
5Department of Physics, The National Institute of Engineering (NIE), Mysore-570008, Karnataka, India.
6Department of Chemistry, College of Education, University of Thamar, Yemen.
Corresponding Author E-mail: malghorbani@yahoo.com
DOI : http://dx.doi.org/10.13005/ojc/330604
Download this article as:
![]()
A new series of substituted quinoline and pyridine derivatives 6a-h are synthesized by the coupling of hydrazide derivatives 4a-b with substituted carboxylic acids 5a-c in the presence of TBTU as a coupling agent and lutidine as a base. The newly synthesized compounds 6a–l is characterized by analytical NMR and mass spectral data. The newly synthesized compounds are subjected to molecular docking studies for the inhibition of human Aurora A kinase target to evaluate their potential value for the treatment of cancers.
KEYWORDS:Synthesized Compounds; Substituted; Molecula
Introduction
Quinoline nucleus represent a significant class of heterocycles, they occur in various natural products, especially in alkaloids and exhibit exceptionally broad spectrum of biological activities [1-3]. The synthesis of quinoline and their derivatives has attracted considerable attention from organic and medicinal chemists for many years as a large number of natural and synthetic products contain this heterocyclic nucleus [4–8]. Some derivatives containing quinoline ring system have been shown to possess useful biological activities, such as antiprion [4], antimicrobial [5-8], antibacterial [9], antitubercular [10- 12] and anticancer activities [13-15]. Similarly, it is well documented that pyridine nucleus is associated with a variety of pharmacological actions, including, pathogenesis of cancer, HIV, autoimmune and inflammatory diseases. [16,17]
In view of the interest in the activity spectrum and profile of quinoline and pyridine, and in continuation of our work on the synthesis of new compounds of pharmacological and biological interest [18-22], we describe herein the preparation and Doking study of some new quinoline and pyridine derivatives (6a-h)
Material an Methods
General Methods
Chemicals were purchased from Sigma Aldrich Chemical Co. TLC was performed on aluminium-backed silica plates and visualized by UV-light. Melting points were determined on a Thomas Hoover capillary melting point apparatus with a digital thermometer. IR spectra were determined on JASCO FT-IR 4000 spectrophotometers. NMR spectra were recorded on Agilent-400 NMR MHz spectrometer in CDCl3. Chemical shifts were recorded in parts per million downfield from tetramethylsilane. Mass spectra were carried out on LC-MS/MS (API-4000) mass spectrometer. The elemental analysis of the compounds was performed on a Perkin Elmer 2400 Elemental Analyzer. The results of elemental analyses were within ±0.4% of the theoretical values.
Chemical Synthesis
General Synthesis of Ethyl Acetate Analogues (3a-b)
A mixture of commercially available 8-hydroxy quinoline or 5-methyl-pyridin-2-ol 1a-b (0.03 mol) and chloroethyl acetate 2 (0.045 mol) in dry acetone (30 ml) with anhydrous potassium carbonate (0.045 mol) were refluxed for 10-16 hours. The reaction mixture was cooled and solvent removed by distillation. The residual mass was triturated with cold water to remove potassium carbonate, and extracted with ether (3×30 ml). The ether layer was washed with 10% sodium hydroxide solution (3×30 ml) followed by water (3×30 ml) and then dried over anhydrous sodium sulphate and evaporated to afford compounds 3a-c. The physical and analytical data of the synthesized title compounds 3a-c are given as follows.
4-(Quinolin-8-yloxy)-acetic Acid Ethyl Ester (3a)
Yield 90%, yellow liquid, IR (cm−1): 1735 (C=O), 3115-3225 (NHNH2);1H NMR (CDCl3): δ (ppm) = 2.36 (t, 3H, CH3), 3.28 (q, 2H, CH2), 4.21 (t, 2H, OCH2), 7.18-8.85 (m, 6H, Ar-H). LC-MS m/z =232 Anal. Cal. for C13H13NO3:C, 67.52; H, 5.67. Found: C, 67.35; H, 5.51%.
(5-Methyl-pyridin-2-yloxy)-acetic Acid Ethyl Ester (3b)
Yield 87%, yellow liquid, IR (cm−1): 1745 (C=O); 3110-3215 (NHNH2)1H NMR (CDCl3): δ (ppm) = 2.27 (t, 3H, CH3), 2.45 (t, 3H, CH3), 3.22 (q, 2H, CH2), 4.29 (t, 2H, OCH2), 7.27-8.74 (m, 3H, Ar-H). LC-MS m/z =196 Anal. Cal. for C10H13NO3: C, 61.53; H, 6.71. Found: C, 61.38; H, 6.54%.
General synthesis of Hydrazide Derivatives (4a-b)
Hydrazine hydrate (0.018 mol) was added to a solution of compounds 3a-b (0.018mol) in absoluteethanol (30 ml) and continuously stirred for 2-4 h at room temperature. A solid was separated out, which was quenched with water (50 ml), filtered and washed with water (50 ml). Finally, the solid was dried under vacuum and recrystallized with ethanol to obtain compounds 4a-b.
(Quinolin-8-yloxy)-acetic Acid Hydrazide (4a)
Yield 80%, white solid mp; 145-147oC, IR (cm−1): 1720 (C=O); 1H NMR (CDCl3): δ (ppm) = 4.45 (s, 2H, NH2), 4.71 (s, 2H, CH2), 7.21-8.87 (m, 6H, Ar), 9.41 (s, 1H, NH), LC-MS m/z =182 Anal. Cal. for C8H11N3O2: C, 53.03; H, 6.12. Found: C, 52.86; H, 6.03%.
(5-Methyl-pyridin-2-yloxy)-acetic Acidhydrazide (4b)
Yield 87%, white solid mp; 178-180oC, IR (cm−1): 1735(C=O); 1H NMR (CDCl3): δ (ppm) =4.23 (s, 2H, NH2), 4.83 (s, 2H, CH2), 7.55-8.37 (m, 3H, Ar), 9.21 (s, 1H, NH); LC-MS m/z =218 Anal. Cal. for C11H11N3O2: C, 60.82; H, 5.10. Found: C, 60.60; H, 5.02%.
General Synthesis of Compounds (6a-h)
Hydrazide derivatives (4a-b, 3 mmol) in dry DCM (20 ml) was stirred at 25–30 oC, and then lutidine (4 mmol) was added, followed by the addition of substituted amino-4-phenyl-1,3-thiazoles (3 mmol). The reaction mixture was stirred at the same temperature for 30 min, then cooled to0–5 oC and TBTU (3 mmol) was added over a period of 30 min maintaining the temperature below 5 oC. The reaction mass was stirred overnight and monitored by TLC using chloroform: methanol (9:1) as the mobile phase. The solvent was evaporated at reduced pressure, quenched by the addition of crushed ice and the obtained solid was filtered, dried and recrystallized from ethanol to afford compounds 6a-h in good yield.
(4-Fluoro-phenyl)-acetic acid N’-[2-(quinolin-8-yloxy)-acetyl]-hydrazide (6a)
Yield 76%, white solid mp; 255-257oC, IR (cm−1) 1640, 1675 (C=O), 3245–3470 (NH–NH); 1H NMR (CDCl3): δ (ppm) = 3.21 (s, 2H, CH2), 4.76 (s, 2H, CH2), 7.26-8.94 (m, 10H, Ar), 10.23 (d, 2H, 2NH); LC-MS m/z =354 Anal. Cal. for C19H16FN3O3: C, 64.58; H, 4.56. Found: C, 64.37; H, 4.80%.
(4-Chloro-phenyl)-acetic acid N’-[2-(quinolin-8-yloxy)-acetyl]-hydrazide (6b)
Yield 80 %, white solid, mp; 258-260oC, IR (cm−1): 1630, 1685 (C=O), 3255–3475 (NH–NH); 1H NMR (CDCl3): δ (ppm) = 3.44 (s, 2H, CH2), 4.83 (s, 2H, CH2), 7.21-8.85 (m, 10H, Ar), 10.24 (d, 2H, 2NH), LC-MS m/z =370 Anal. Cal. for C19H16ClN3O3: C, 61.71; H, 4.36. Found: C, 61.66; H, 4.17%.
(3,4-Dichloro-phenyl)-acetic acid N’-[2-(quinolin-8-yloxy)-acetyl]-hydrazide (6c)
Yield 73%, white solid mp; 228-230oC, IR (cm−1): 1660, 1690 (C=O), 3235–3455 (NH–NH); 1H NMR (CDCl3): δ (ppm) = 3.35 (s, 2H, CH2), 4.69 (s, 2H, CH2), 7.43-8.97 (m, 9H, Ar), 10.21 (d, 2H, 2NH). LC-MS m/z =405 Anal. Cal. for C19H15Cl2N3O3: C, 56.45; H, 3.74. Found: C, 56.32; H, 3.51%.
4-Methoxy-benzoic acid N’-[2-(quinolin-8-yloxy)-acetyl]-hydrazide (6d)
Yield 78%, white solid mp; 253-255oC, IR (cm−1):1635, 1680 (C=O), 3230–3470 (NH–NH); 1H NMR (CDCl3): δ (ppm) = 3.30 (s, 3H, CH3), 4.88 (s, 2H, CH2), 7.37-8.74 (m, 10H, Ar), 10.27-10.34 (d, 2H, 2NH). LC-MS m/z = 352 Anal. Cal. for C19H17N3O4: C, 64.95; H, 4.88. Found: C, 64.81; H, 4.67%.
(4-Fluoro-phenyl)-acetic acid N’-[2-(5-methyl-pyridin-2-yloxy)-acetyl]-hydrazide (6e)
Yield 71%, white solid mp; 211-213oC, IR (cm−1):1640, 1695 (C=O), 3250–3475 (NH–NH); 1H NMR (CDCl3): δ (ppm) = 1.89 (s, 3H, CH3), 3.34 (s, 2H, CH2), 4.41 (s, 2H, CH2), 6.18-7.69 (m, 7H, Ar), 10.13 (d, 2H, 2NH). LC-MS m/z = 318 Anal. Cal. for C16H16FN3O3: C, 60.56; H, 5.08. Found: C, 60.45; H, 5.04%.
(4-Chloro-phenyl)-acetic acid N’-[2-(5-methyl-pyridin-2-yloxy)-acetyl]-hydrazide (6f)
Yield 65%, white solid mp; 282-284oC, IR (cm−1): 1635,1685(C=O), 3240–3460 (NH–NH); 1H NMR (CDCl3): δ (ppm) = 1.93 (s, 3H, CH3), 3.51 (s, 2H, CH2), 4.68 (s, 2H, CH2), 6.28-7.65 (m, 7H, Ar), 10.32 (d, 2H, 2NH). LC-MS m/z = 334 Anal. Cal. for C16H16ClN3O3: C, 57.58; H, 4.83. Found: C, 57.67; H, 4.35%.
(3,4-Dichloro-phenyl)-acetic acid N’-[2-(5-methyl-pyridin-2-yloxy)-acetyl]-hydrazide (6g)
Yield 68%, white solid mp; 286-288oC, IR (cm−1): 1645, 1690 (C=O), 3230–3455 (NH–NH); 1H NMR (CDCl3): δ (ppm) = 1.98 (s, 3H, CH3), 3.47 (s, 2H, CH2), 4.50 (s, 2H, CH2), 6.27-7.55 (m, 6H, Ar), 10.20 (d, 2H, 2NH).13 C NMR (100MHz, CDCl3): 34.4, 54.19, 126.24, 133.49, 136.74, 137.65, 138.87, 143.36, 164.15. LC-MS m/z = 368 Anal. Cal. for C16H15Cl2N3O3: C, 52.19; H, 4.11. Found: C, 52.23; H, 4.06%.
4-Methoxy-benzoic acid N’-[2-(5-methyl-pyridin-2-yloxy)-acetyl]-hydrazide (6h)
Yield 64%, white solid mp; 274-276oC, IR (cm−1): 1625, 1675 (C=O), 3240–3460 (NH–NH); 1H NMR (CDCl3): δ (ppm) = 2.08 (s, 3H, CH3), 3.24 (s, 3H, CH3), 4.47 (s, 2H, CH2), 6.33-7.70 (m, 7H, Ar), 10.28 (d, 2H, 2NH). LC-MS m/z = 316 Anal. Cal. for C16H17N3O4: C, 60.94; H, 5.43. Found: C, 61.01; H, 5.32%.
Result and Discussion
Synthesis of the target compounds 6a-h was performed according to the reactions illustrated in scheme 1. Initially, esterification of 8-hydroxy quinoline and 5-methyl-pyridin-2-ol 1a-b was performed by using chloroethyl acetate 2 in the presence of anhydrous potassium carbonate and dry acetone at reflux temperature to afford compounds 3a-b. These materials were stirred with hydrazine hydrate in ethanol to afford hydrazide derivatives 4a-b. Finally, the target derivatives 6a-h were obtained in good yields by stirring of compounds 4a-b with substituted carboxylic acids 5a-c in the presence of N,N,N’,N’-tetramethyl-o-(benzotriazol-1-yl) uranium tetrafluoroborate (TBTU) as a coupling agent and lutidine as a base in dry DCM at RT. The structure elucidations of the synthesized compounds were confirmed by IR, NMR, mass spectrometry and elemental analysis.
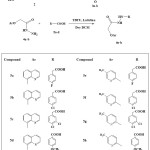 |
Scheme 1: Synthesis of quinoline and pyridine derivatives (6a-h) Click here to View scheme |
Supplementary Material
The spectra of the title compound 4a were considered as a representative example of the series 4a-b. In IR spectra, the compound 4a showed band at 1720 and in between 3115-3225 cm-1 corresponding to carbonyl and NH-NH2 stretching frequencies respectively. In 1H NMR spectra of compound 4a showed one singlet at d 4.45 assigned to NH2 and broad singlet at d 4.71 assigned to OCH2 protons, it also revealed broad singlet at d 9.41 assigned to amide proton as well as multiplet signals appeared in the range d 7.21-8.87 for aromatic protons.
In IR spectra of compounds 6a was confirmed by the appearance of one more carbonyl at 1685 cm-1 and disappearance of the NH2 absorption peak. In addition, 1H NMR spectra showed disappearance of NH2 protons at 4.45 and an increase in one more NH proton and four aromatic protons with earlier aromatic proton peaks at d10.23 and 7.26-8.94 respectively, which clearly evidence the formation of compound 6a. The mass spectra of compound 6a gave significant stable (M+) peak at m/z 354. Further, all the target compounds 6a-h were clearly confirmed by 13C NMR.
Molecular Docking Studies
The molecular docking was performed and analyzed using Auto Dock 4.2. A Lamarkian genetic algorithm method implemented in the program suite was employed to identify appropriate binding modes and conformation of the ligand molecules. Gasteiger charges were added and the rotatable bonds were set by the Auto Dock tools and all torsions were allowed to rotate. Polar hydrogen atoms were added and Kollaman charges were assigned to the protein using Auto Dock tools (ADT). All the compounds (6a-h) were found to have minimum binding energy ranging from -6.29 to -8.20 kJ/mol with Aurora A kinase (PDB Code:3FDN).
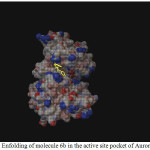 |
Figure 1: Enfolding of molecule 6b in the active site pocket of Aurora Kinase Click here to View figure |
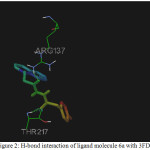 |
Figure 2: H-bond interaction of ligand molecule 6a with 3FDN |
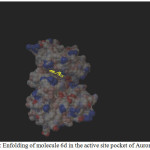 |
Figure 1: Enfolding of molecule 6d in the active site pocket of Aurora Kinase |
 |
Figure 2: H-bond interaction of ligand molecule 6c with 3FDN |
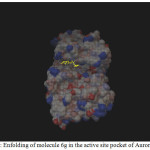 |
Figure 1: Enfolding of molecule 6g in the active site pocket of Aurora Kinase |
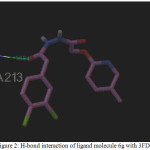 |
Figure 2: H-bond interaction of ligand molecule 6g with 3FDN |
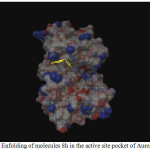 |
Figure 1: Enfolding of molecules 8h in the active site pocket of Aurora Kinase |
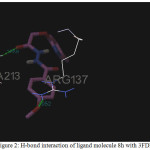 |
Figure 2: H-bond interaction of ligand molecule 8h with 3FDN |
Table 1: The dock score results of the different quinolinederivativeswith Aurora A kinase target (PDB Code: 3FDN)
|
Compounds |
Binding Energy (kJ mol-1) |
Ligand Efficiency |
Inhibition Constant |
vd W+H-bond+desolv energy |
No. of H- bonds |
Bonding residues |
Bond Length (Å) |
|
6a |
-6.96 |
-0.27 |
7.97 |
-7.9 |
2 |
3FDN:A:ARG137:HH22 |
1.958 |
|
6b |
-7.73 |
-0.3 |
2.15 |
-9.04 |
– |
– |
– |
|
6c |
-8.2 |
-0.3 |
978.84 |
-9.12 |
1 |
3FDN:A: LYS162:HZ3 |
2.155 |
|
6d |
-7.64 |
-0.29 |
2.51 |
-8.99 |
– |
– |
– |
|
6e |
-6.29 |
-0.27 |
24.63 |
-6.93 |
1 |
3FDN:A: LYS162:HZ3 |
1.778 |
|
6f |
-6.67 |
-0.29 |
12.98 |
-8.25 |
1 |
3FDN:A: LYS162:HZ3 |
2.171 |
|
6g |
-7.31 |
-0.3 |
4.36 |
-8.48 |
1 |
3FDN:A: ALA213:HN |
1.884 |
|
8h |
-6.63 |
-0.29 |
13.9 |
-7.99 |
2 |
3FDN:A:ARG137:HH22| |
2.052 |
Among the molecules tested for docking study, (3,4-Dichloro-phenyl)-acetic acid N’-[2-(quinolin-8-yloxy)-acetyl]-hydrazide (6c) showed minimum binding energy of -8.20 kJ/mol with ligand efficiency of -0.30.Most of the residues that are in close proximity to the inhibitor are hydrophobic in nature. The ligand molecules, 6b, 6d and 6g revealed binding energy of -7.73, -7.64 and -7.31 kJ/mol, with ligand efficiency of -0.30, -0.29 and -0.30, respectively. These molecules were completely wrapped by active site amino acid residues at the active site pocket region (Fig. 1). Similarly, molecules 6a, 6c and 6g were found to show hydrogen bond interaction with active site amino acid residues Arg 137, Thr 217, Lys 162, Ala 213 and at a distance of (1.958 and 1.778), (2.155) and (1.884) Å, respectively (Fig. 2).The docking study results showed that the molecules (6a-h) have good inhibition constant, vdW + Hbond + desolv energy with best RMSD value.The details of docked score results of the molecules with Aurora A kinase (PDB Code:3FDN) are given in the Table 1 (see supplementary material).
References
- Embrey, M.W.; Wai, J.S.; Funk, T.W.; Homnick, C.F.; Perlow, D.S.; Young, S.D.; Vacca, J.P.; Hazuda, D.J.; Felock, P.J.; Stillmock, K.A.; Witmer, M.V.; Moyer, G.; Schleif, W.A.; Gabryelski, L.J.; Jin, L.; Chen, IWu.; Ellis, J.D.; Wong, B.K.; Lin, J.H.; Leonard, Y.M.; Tsou N.N.; Zhuang, L. Bioorg. Med. Chem. Lett. 2005, 15, 4550-4554.
CrossRef - Zhuang, L.; Wai, J.S.; Embrey, M.W.; Fisher, T.E.; Egbertson, M.S.; Payne, L.S Guare, J. P.; Vacca, J.P.; Hazuda, D.J.; Felock, P.J.; Wolfe, A.L.; Stillmock, K.A.; Witmer, M.V.; Moyer, G.; Schleif, W.A.; Gabryelski, L.J.; Leonard, Y.M.; Lynch, J.J.; Michelson, S.R.; Young, S.D. J. Med. Chem. 2003, 46, 453-456.
CrossRef - Atanasova, M.; Ilieva, S.; Galabov, B. Euro. J. Med. Chem. 2007, 42, 1184-1192.
CrossRef - Cope H, Mutter R, Heal W, Pascoe C, Brown P, Pratt S, Chen. B. Eur J Med Chem. 2006; 41: 1124-1143.
CrossRef - Mamladesai, S.N.; Katagi, M.S.; Khare, P.V.; Maddi, V.S.; Bhat, A.R. Indian J. Hetero. Chem. 2008, 17, 381-382.
- Kamel, M.A.; Lobna, A.M.; El-Sayed, L.M.; Mohamed, H.I.; Rania B.H. Biorg Med. Chem. 2006, 14, 8675-8682.
CrossRef - Ola, E.I.; Sayed, A.; Fatma, E.M.; Shada, E.I.; Badr, A.A.; Maher, H.E. Indian J. Hetero. Chem. 2005, 14, 327-330.
- Kansagra, B.P.; Bhatt, H.H.; Parikh, A.R. Indian J. Hetero. Chem. 2000, 10, 05-08.
- Santhosh Kumar, S.; Ajay, N.; Tiwari, R.P.; Praveen. J. Indian J. Chem. 2006, 45, 1734-1739.
- Amit, N.; Rahul, J. Indian J. Chem. 2008, 47, 117-128.
- Vangapandu, S.; Jain, M.; Jain, R.; Kaur, S.; Singh, P.P. Bioorg Med Chem. 2004, 12, 2501- 2508.
CrossRef - Vikramdeep, M.; Amit, N.; Balasubramanian, V.; Prakash, P.B.; Sarbjit, S.J.; Sukhraj K, Prati, P. S.; Rahul, J. Biorg Med. Chem. 2004, 12, 6465-6472.
CrossRef - Tseng, C.H.; Chen, Y.L.; Lu, P.J.; Yang, C.N.; Tzeng, C.C. Bioorg Med Chem. 2008, 16, 3153-3162.
CrossRef - Katarzyna, S.; Lukasz, K.S. Wojciech, S.J.; Joanna, W.; Marta, S.; Wanda, P.C. Exper. Therp. 2007, 61, 41-200.
- Zhao, Y.L.; Chen, Y.L.; Chang, F.S.; Tzeng, C.C.; Eur J Med Chem. 2005, 40, 792-797.
CrossRef - Lazennec, G.; Richmond, A. Trends Mol Med. 2010, 16, 133–144.
CrossRef - Schwarz, M.K.; Wells, T.N. Nat Rev Drug Discov. 2002, 1, 347–358.
CrossRef - Al-Ghorbani, M.; Prabhu, T.; Gurupadaswamy, H.D.;Vigneshwaran, V.; Prabhakar. B.T.; Khanum, S. A. Bioorg. Chem, 2017, 71, 55-66.
CrossRef - Al-Ghorbani, M.; Prabhu, T.;, Vigneshwaran, V.; Gurupadaswamy, H.D.; Girish, V.; Prabhakar, B.T.; Khanum, S.A. Bioorg. Chem, 2016, 65, 73–81.
CrossRef - Al-Ghorbani, M.; Pavankumar, G.S.; Naveen, P.; Shamanth, H.G, Prabhakar, B.T.; Shaukath Ara Khanum, Bioorg. Chem, 65, 2016, 110–117.
CrossRef - Al-Ghorbani, M.; Vigneshwaran, V.; Lakshmi Ranganatha, V.; Prabhakar, B.T.; Khanum, S.A. Bioorg. Chem, 2015, 60, 136-146.
CrossRef - Al-Ghorbani M, Rekha N.D, Lakshmi Ranganatha V, Prashanth V, Veerabasappagowda T, Khanum S.A, Russian J. Bioorg. Chem, 2015, 41, 4, 1–8.







ISSN Online: 2231-5039

![]()




{kind=link}