Studies of Stereo-Selective Cyclo-Additions and Transformations of Substituted 2-Cyclopenten-1-one with Chiral Anthracene Templates.
Weerachai Phutdhawong1, Gedsirin Eksinitkun1,2, Yonlada Jaroensuk2, Anthony C. Willis3 and Waya S. Phutdhawong2
1Department of Chemistry, Faculty of Liberal Arts and Science, Kasetsart University, Kamphaeng Saen Campus, Nakhon Pathom, 73140, Thailand.
2Department of Chemistry, Faculty of Science, Silpakorn University, Nakhon Pathom, 73000, Thailand.
3Research School of Chemistry, Australian National University, Canberra, A. C. T. 0200, Australia.
Corresponding Author E-mail: waya.sengpracha@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/330601
The chiral (S)-9-(1-methoxyethyl), (R)-9-(1,2-dimethoxyethyl) and 9-(1R, 2R)-(1,2-dimethoxypropyl) anthracenes were synthesised and used for the thermal Diels-Alder reaction with cyclopentene-3,5-dione. Unlike the maleic anhydride and N-substituted malemides, the cyclo-adducts were obtained with high regio-selectivity as a single diastereomer. The X-ray structure of the cyclo-adduct showed an enol form but the 13C NMR showed resonances for two cyclopentanone carbonyl groups suggesting the solution structure is in the diketone form. Stereo-controlled studies using organomagnesium additions to the carbonyl groups resulted in hydrolytic cleavage of the enol ether and elimination of water to give β-alkylketone anthracene adducts. These were unsuccessful in preparing chiral cyclopentenone core structures.
KEYWORDS:Cyclopentenone; Cyclopentene-3,5-Dione; Chiral Anthracene; Organomagnesium
Download this article as:| Copy the following to cite this article: Phutdhawong W, Eksinitkun G, Jaroensuk Y, Willis A. C, Phutdhawong W. S. Studies of Stereo-Selective Cyclo-Additions and Transformations of Substituted 2-Cyclopenten-1-one with Chiral Anthracene Templates. Orient J Chem 2017;33(6). |
| Copy the following to cite this URL: Phutdhawong W, Eksinitkun G, Jaroensuk Y, Willis A. C, Phutdhawong W. S. Studies of Stereo-Selective Cyclo-Additions and Transformations of Substituted 2-Cyclopenten-1-one with Chiral Anthracene Templates. Orient J Chem 2017;33(6). Available from: http://www.orientjchem.org/?p=40159 |
Introduction
The cyclopentenone skeleton has been reported in diverse biological active compounds. For example, prostanoids such as clavulone I and clavulone II1 isolated from marine natural products and exhibiting strong cytotoxicity. Untenone A2 isolated from the Okinawan marine sponge Plakortis sp. which inhibites cell proliferation of L1210 leukaemia (IC50 = 0.4 µg/mg) and mammalian DNA polymerases (pol. α and β), and human terminal deoxynucleotidyl transferase (TdT).3 Recently, TEI-9826,4 an antitumor agent in preclinical trials, has also been prepared.
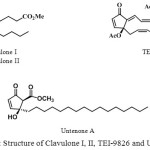 |
Figure 1: Structure of Clavulone I, II, TEI-9826 and Untenone A |
Many strategies have been developed to synthesise cyclopentenones including the Nazarov cyclisation,5 the Pauson-Khand reaction,6 metal-catalysed cyclisation,7 and Diels-Alder or retro Diels-Alder reactions using anthracene.8 Thus, the synthesis of the cyclopentenone ring system is highly desirable, particularly with control of relative and absolute stereo-chemistry. The Diels-Alder/retro Diels-Alder reactions between 9-substituted chiral anthracene and maleic anhydride and N-substituted maleimides, or p-benzoquinone has been described in previous reports as chiral anthracene could stereo-control the cyclo-addition and control asymmetric substitutions at the carbonyl groups.9-11 In addition, regio- and diastereoselective additions has also been achieved in the Diels-Alder reaction of 2-cyclopentene-1-one and chiral anthracene templates.12 The work of the authors in the preparation of (S)-9-(1-methoxyethyl)anthracene, (R)-9-(1,2-dimethoxyethyl)anthracene and 9-(1R, 2R)-(1,2-dimethoxypropyl)anthracene, and the availability of cyclopentene-3,5-dione prompted an investigation of the stereo-selective Diels-Alder reactions and transformation of cyclo-adducts via regio-selective and stereo-selective manipulations prior to the asymmetric synthesis of bioactive cyclopentenones.
The Diels-Alder reaction of cyclopentene-3,5-dione, a good dienophile, and anthracene has been reported to give a completely enolic anthracene adduct after refluxing in benzene for four days.13 Thus, it was considered that the reaction of cyclopentene-3,5-dione with our synthetic chiral anthracenes might provide single diastereomers of corresponding enolic anthracene adducts. The single diastereomer could be obtained as the hydrogen bond interaction between the enol oxygen and the anthracene C-9 hydrogen similar to the previous discussion in the cyclo-addition of chiral anthracene and maleic anhydride or N-methyl maleimide.9 The stereo-selective substitution from the less hindered face might provide asymmetric synthesis of cyclopentenone derivatives.
Experimental
General Methods
Melting points were determined with a Stuart Scientific SMP 2 melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded in CDCl3 with a Bruker Avance 300 spectrometer (300 MHz for 1H, 75 MHz for 13C) using TMS as an internal standard. Mass spectra were recorded with a POLARIS Q or HEWLETT PACKARD 5973 mass spectrometer. Reactions were monitored by TLC using aluminium or plastic sheets pre-coated with silica gel 60 F254. Column chromatography was performed with Kieselgel 60.
9-Vinylanthracene (1) was prepared as described previously14, (S)-9-(1-methoxy ethyl)anthracene (6) was synthesised as described previously using Snyder’s method15, and cyclopentene-3,5-dione (7) was commercially available.
Synthesis Method
(R)-9-(1,2-dihydroxyethyl)anthracene (2a)
A mixture of 9-vinylanthracene 0.1 g (1a) (0.49 mmol, 1.0 equiv), AD-mix β (0.69 g, ratio 1.414 g : 1.0 mmol) and methansulfonamide 0.1 g in tert-butanol (ratio 1:1 H2O : tert-butanol) 7 mL was stirred at 0ºC in a cool room for four days. Approximately half a teaspoon Na2SO3 was then added to the mixture with a further 30 min stirring, followed by extraction with CH2Cl2 (3×20 mL). The combined organic phase was dried over anh. Na2SO4, filtered and concentrated in vacuo. Purification of the residue by flash column chromatography (silica gel) with Hexane/EtOAc (1 :1) as the mobile phase gave a light yellow solid (2a) 0.08 g (0.34 mmol, 70%); m.p. 130-132ºC (lit.16 133.5ºC),
![]()
(c 0.22, EtOH); Rf = 0.34); 1H-NMR (300 Hz, CDCl3) δ 8.61 (2H, d, J = 9.0 Hz, ArH), 8.38 (1H, s, ArH), 7.97 (2H, d, J = 9.0 Hz, ArH), 7.48-7.40 (4H, m, ArH), 6.32 (1H, dd, J = 4.0, 10.0 Hz, CH), 4.42 (1H, dd, J = 10.0, 12.0 Hz, CH), 3.86 (1H, dd, J = 4.0, 12.0 Hz, CH); 13C NMR (75 MHz, CDCl3) δ 60.4, 66.2, 124.7, 124.9, 125.9, 128.7, 129.3, 129.9, 130.4, 131.9.
(1R,2R)-1-(anthracen-9-yl)propane-1,2-diol (2b)
A mixture of (E)-9-(prop-1-en-1-yl)anthracene (1b) 0.1 g (0.458 mmol, 1.0 equiv), AD-mix β (0.66 g, ratio 1.414 g : 1.0 mmol) and methansulfonamide 0.1 g (1.05 mmol, 1.0 equiv) in tert-butanol (ratio 1:1 H2O : tert-butanol) 7 mL was stirred at 0ºC in a cool room for four days. Approximately half a teaspoon Na2SO3 was then added to the mixture with a further 30 min stirring, followed by extraction with CH2Cl2 (3×20 mL). The combined organic phase was dried over anh. Na2SO4, filtered and concentrated in vacuo. Purification of the residue by flash column chromatography (silica gel) with Hexane/EtOAc (1 :1) as the mobile phase gave a light yellow solid (2b) 0.09 g (0.36 mmol, 45%); m.p. 130-132ºC,
![]()
(c 0.75, CHCl3); 1H-NMR (300 Hz, CDCl3) δ 8.65 (2H, brs, ArH), 8.41 (1H, s, ArH), 8.01-7.97 (2H, m, ArH), 7.48-7.44 (4H, m, ArH), 5.95 (1H, d, J = 9.0 Hz, ArH), 4.83-4.73 (1H, m, CH), 0.84 (3H, d, J = 6.0 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 19.2, 71.1, 75.6, 124.8, 125.8, 128.6, 129.3, 130.2, 131.4, 131.7; HR-ESI MS calculated for C17H16NaO2 (M+Na)+ 275.1048, found 275.1043.
Methylation of (R)-9-(1,2-dihydroxyethyl)anthracene (2a)
A mixture of (R)-9-(1,2-dihydroxyethyl)anthracene (2a) 0.1 g (0.42 mmol, 1.0 equiv) with sodium hydride 60% wt (0.07 g, 3.0 mmol) was stirred in 15 mL dry THF under argon at 0ºC for 10 min. methyl iodide (0.13 mL, 2.00 mmol) was added dropwise and the mixture was stirred at room temperature for 6 hours. The resulting mixture was quenched with an aqueous saturated NH4Cl solution, extracted with Et2O (3×20 mL) and washed with water (30 mL) and saturated NaCl (30 mL). The combined organic layer was dried (anh. Na2SO4), filtered and concentrated in vacuo. Purification of the residue by preparative layer chromatography using Hexane/EtOAc (10:1) as the mobile phase gave the first fraction at Rf= 0.45 as a yellow solid of (R)-9-(1,2-dimethoxyethyl)anthracene (3a) 0.06 g (0.21 mmol, 52%) and the second fraction at Rf= 0.15 as a yellow oil of (R)-1-(anthracen-9-yl)-2-methoxyethanol (4a) 0.04 g (0.16 mmol, 36%). (R)-9-(1,2-dimethoxyethyl)anthracene (3a); m.p. 78.0-80.0ºC (lit.17 78.0-80.0ºC);
![]()
(c 0.75, CHCl3); 1H-NMR (300Hz, CDCl3) δ 8.70 (2H, brs, ArH), 8.44 (1H, s, ArH), 8.02 (2H, d, J = 12.0 Hz, ArH), 7.52-7.45 (4H, m, ArH), 6.05 (1H, dd, J = 3.0, 9.0 Hz, CH), 4.34 (1H, dd, J = 9.0, 12.0 Hz, CH), 3.65 (1H, dd, J = 3.0, 9.0 Hz, CH), 3.47 (3H, s, OCH3), 3.29 (3H, s, OCH3); 13C-NMR (75 MHz, CDCl3) δ 56.9, 59.3, 75.9, 80.0, 124.9, 125.9, 127.2, 128.6, 128.9, 129.3, 130.5, 131.5; HR-ESI MS calculated for C18H18O2 (M+Na)+ 289.1204, found 289.1199.
(R)-1-(anthracen-9-yl)-2-methoxyethanol (4a);
![]()
(c 0.22, CHCl3)); 1H-NMR (300Hz, CDCl3) δ 8.70 (2H, d, J = 9.0 Hz, ArH), 8.43 (1H, s, ArH), 8.00 (2H, d, J = 9.0 Hz, ArH), 7.53-7.43 (4H, m, ArH), 6.49 (1H, dd, J = 3.0, 9.0 Hz, CH), 4.28 (1H, dd, J = 9.0, 9.0 Hz, CH2), 3.68 (1H, dd, J = 3.0, 9.0 Hz, CH2), 3.53 (3H, s, OCH3); 13C-NMR (75 MHz, CDCl3) δ 48.9, 57.2, 68.6, 122.8, 123.8, 125.3, 126.7, 127.3, 127.9, 129.7, 132.2; HR-EI MS calculated for C17H16O2 (M)+ 252.1151, found 252.1150.
9-((1R,2R)-1,2-dimethoxypropyl)anthracene (3b)
A mixture of (1R,2R)-1-(anthracen-9-yl)propane-1,2-diol (2b) 0.1 g (0.396 mmol, 1.0 equiv) with sodium hydride 0.02 g (0.793 mmol, 2.0 equiv) was stirred in dry THF 15 mL under argon at 0 ºC for 10 minutes. Methyl iodide (0.18 mL, 2.00 mmol) was added dropwise and the mixture was stirred at room temperature for 6 hours. The resulting mixture was quenched with an aqueous saturated NH4Cl solution, extracted with Et2O (3×20 mL) and washed with water (30 mL) and saturated NaCl (30 mL). The combined organic layer was dried (anh. Na2SO4), filtered and concentrated in vacuo. Purification of the residue by preparative layer chromatography using hexane/EtOAc (4:1) as the mobile phase gave compound (3b) as a yellow solid 0.02 g (0.07 mmol, 18% ); m.p. 110-112ºC;
![]()
(c 0.75, CHCl3); 1H-NMR (300Hz, CDCl3) δ 9.02 (1H, m, ArH), 8.44 (2H, s, ArH), 8.02 (2H, d, J = 6.0 Hz, ArH), 7.55-7.44 (4H, m, ArH), 5.75 (1H, d, J = 9.0 Hz, CH), 4.43-4.36 (1H, m, CH), 3.61 (3H, s, OCH3), 3.24 (3H, s, OCH3), 0.68 (3H, d, J = 6.0 Hz, CH3); 13 C-NMR (75 MHz, CDCl3) δ 16.3, 56.9, 57.6, 80.2, 84.3, 123.5, 124.7, 125.1, 125.4, 126.3, 126.6, 127.3, 128.5, 129.0, 129.5, 130.0, 131.8, 134.1; HR-ESI MS calculated for C19H20NaO2 (M+Na)+ 303.1361, found 303.1348.
(R)-1-(Anthracen-9-yl)-2-methoxyethyl-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate (5a)
To a solution of (R)-1-(anthracen-9-yl)-2-methoxyethanol (4a) 60 mg (0.237 mmol, 1.0 equiv.) in CH2Cl2 (15 mL), (S)-(-)-α-methoxy-α-(trifluoromethyl)phenylacetic acid (S-Mosher) 110 mg (0.470 mmol, 2 equiv.), N,N-dicyclohexylcarbodiimide (DCC) 97.8 mg (0.474 mmol, 2 equiv) and 4-dimethylamino pyridine (DMAP) 3.5 mg (0.0286 mmol, 0.12 equiv) were added and stirred at room temperature overnight. The reaction mixture was filtered and concentrated. Purification by preparative layer chromatography using hexane/EtOAc (10:1) as the mobile phase gave a light yellow solid (5a) 0.32 g (0.68 mmol, 29%); 1H-NMR (300 Hz, CDCl3) δ 8.70 (2H, brs, ArH), 8.48 (1H, s, ArH), 8.04 (2H, d, J = 9.0 Hz, ArH), 7.61-7.38 (9H, m, ArH), 6.10 (1H, dd, J = 6.0, 9.0 Hz, CH), 5.23 (1H, dd, J = 12.0, 12.0 Hz, CH), 4.62 (1H, dd, J = 3.0, 12.0 Hz, CH), 3.57 (3H, s, CH3), 3.22 (3H, s, CH3); 13 C-NMR (75 MHz, CDCl3) δ 45.3, 55.1, 63.7, 100.0, 122.8, 123.8, 124.1, 125.3, 126.7, 127.3, 127.5, 127.9, 128.1, 128.5, 129.1, 129.7, 131.9, 132.2, 165.7; HR-ESI MS calculated for C27H23F3NaO4 (M+Na)+ 491.1446, found 491.1452.
(9R,10S,11S,12S)-10-((R)-1,2-dimethoxyethyl)-15-hydroxy-10,11-dihydro-9H-9,10-[1,2]epi cyclepentaanthracen-13(12H)-one (8a)
A mixture of (R)-9-(1,2-dimethoxyethyl)anthracene (3a) 0.03 g (0.114 mmol, 1.0 equiv) and 4-cyclopentene-1,3-dione 0.012 g (0.125 mmol, 1.2 equiv) in dry benzene 1 mL under argon was refluxed at 110 °C overnight. The reaction mixture was cooled to room temperature and purification of the residue by preparative-layer chromatography (PLC) (silica gel) using hexane/EtOAc (1:1) as the mobile phase gave (9R,10S,11S,12S)-10-((R)-1,2-dimethoxy ethyl)-15-hydroxy-10,11-dihy-dro-9H-9,10-[1,2]epicyclepentaanthracen-13(12H)-one (8a) as a yellow oil 0.03 g (10% yield); 1H-NMR (300 MHz, CDCl3) δ 10.92 (1H, s, OH), 7.45-7.12 (8H, m, ArH), 5.72 (1H, d, J = 6.0 Hz, CH), 4.88 (1H, s, CH), 4.62 (1H, d, J = 6.0 Hz, CH), 3.56 (3H, s, OCH3), 3.44 (3H, s, OCH3), 3.47 (1H, d, J = 6.0 Hz, CH), 3.28 (1H, dd, J = 9.0 Hz, CH), 2.98 (2H, m, CH); 13C-NMR (75 MHz, CDCl3) δ 50.6, 53.5, 55.4, 58.8, 60.6, 75.0, 76.6, 100.0, 127.4, 127.6, 128.9, 129.3, 129.7, 129.8, 130.5, 142.6, 143.4, 144.3, 147.4, 203.0, 211.9;; HR-ESI MS calculated for C23H22NaO4 (M+Na)+ 385.1416, found 385.1441.
(9R,10S,11S,12S)-10-((1R,2R)-1,2-dimethoxypropyl)-15-hydroxy-10,11-dihydro-9H-9,10-[1,2]epicyclopentaanthracen-13(12H)-one (8b)
A mixture of 9-((1R,2R)-1,2-dimethoxypropyl)anthracene (3b) 0.02 g (0.713 μmol), 4-cyclopentene-1,3-dione 0.01 g (1.5 eq.) and dry benzene 1 ml were added in a pressure tube and heated at 110๐C overnight. After cooling to room temperature, the solvent was evaporated to dryness and the crude product was purified by column chromatography (silica gel, Hexane:EtOAc (1:1)). The product of (9R,10S,11S,12S)-10-((1R,2R)-1,2-dimethoxy propyl)-15-hydroxy-10,11-dihydro-9H-9,10-[1,2]epicyclopentaanthracen-13(12H)-one (8b) was obtained as a yellow viscous oil 0.002 g (11% yield); 1H-NMR (300 MHz, CDCl3) δ 10.46 (1H, s, OH), 7.35 (2H, d, J = 6.0 Hz, ArH), 7.25 (1H,s, ArH), 7.14-7.03 (4H, m, ArH), 6.85 (2H, d, J = 6.0 Hz, ArH), 5.12 (1H, s, CH), 4.70 (1H, s, CH), 4.59 (1H, d, J = 3.0 Hz, CH), 4.13 (1H, q, J = 6.0 Hz, CH), 3.93 (3H, s, OCH3), 3.47 (3H, s, OCH3), 3.18 (1H, d, J = 6.0 Hz, CH), 3.06 (1H, dd, J = 6.0, 6.0 Hz, CH), 1.85 (3H, d, J = 6.0 Hz, CH3); 13C-NMR (75 MHz, CDCl3) δ 19.4, 46.0, 46.3, 50.6, 53.4, 55.8, 60.3, 75.2, 83.5, 111.4, 122.6, 124.2, 124.6, 125.3, 125.6, 126.1, 126.3, 126.5, 141.7, 142.3, 147.3, 144.2, 203.8, 211.9; HR-ESI MS calculated for C24H24NaO4 (M+Na)+ 399.1572, found 399.1600.
15-Hydroxy-10-((S)-1-methoxymethyl)-11,12-dihydro-9H-9,10-[1,2]epicyclopenta-anthracen-13-one (8c)
A mixture of (S)-9-(1-Mehtoxyethyl)anthracene (6) 3.55 g (15.0 mmol, 1 equiv) and cyclopentene-1,3-dione (7) 1.75 g (18.4 mmol, 1.2 equiv) in anhydrous benzene (25 mL) under argon was refluxed at 120 ºC for 6 h. After cooling to room temperature, the precipitated adduct was filtered and purified by flash column chromatography on silica gel (diethyl acetate) to give 15-Hydroxy-10-((S)-1-methoxymethyl)-11,12-dihydro-9H-9,10-[1,2]epicyclopenta-anthracen-13-one (8c) 4.40 g (88%); m.p. 265-267ºC; 1H-NMR (300Hz, CDCl3:MeOD, 9:1) δ 7.86 (1H, m, ArH), 7.35-7.32 (1H, m, ArH), 7.15-7.05 (6H, m, ArH), 5.04 (1H, q, J = 2.1 Hz, CH), 4.54 (1H, d, J = 7.8 Hz, CH), 3.89 (3H, s, OCH3), 3.72 (1H, s, CH), 3.51 (1H, d, J = 6.4 Hz, OH), 3.03 (1H, d, J = 6.2, ArH), 1.94 (3H, d, J = 5.7 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 20.8, 34.7, 50.6, 53.5, 55.4, 60.6, 78.0, 127.4, 127.6, 128.9,129.3, 129.7, 129.8, 130.5, 142.6, 143.4, 144.3, 147.4, 203.8, 211.9; HR-ESI MS calculated for C22H20O3 (M+H)+ 332.1459, found 332.1412.
Methylation of 15-Hydroxy-10-((S)-1-methoxymethyl)-11,12-dihydro-9H-9,10-[1,2]epicy clopenta-anthracen-13-one (8c)
To a solution of 15-Hydroxy-10-((S)-1-methoxymethyl)-11,12-dihydro-9H-9,10-[1,2]epi cyclopenta-anthracen-13-one (8c) 0.23 g (0.69 mmol) and sodium hydride 60 mg (1.50 mmol) in DMF 4 mL at 0 ºC under argon, methyl iodide 0.1 mL (1.40 mmol, 2 equiv) was added dropwise. The mixture was then allowed to warm to room temperature and stirred for 9 hours before being quenched by the addition of saturated ammonium chloride solution (10 mL). After stirring for 15 minutes at room temperature, the mixture was extracted with diethyl ether (3×10 mL), and the combined organic layers were dried over anh. Na2SO4. After filtration and evaporation under reduced pressure, the residue was purified by flash column chromatography on silica gel (Hexane/EtOAc, (2:1)) to give 15-methoxy-10-((S)-1-metho xyethyl)-11,12-dihydro-9H-9,10-[1,2]epicyclo penta-anthracen-13-one (9a) 0.072 g (30%) and 13-methoxy-10-((S)-1-methoxyethyl)-11,12-dihydro-9H-9,10-[1,2]epicyclopenta-anthra cen-15-one (10a) 0.12 g (50%) as a yellow solid. (9a); m.p. 193-195 ºC, 1H-NMR (300Hz, CDCl3) δ 7.79 (1H, m, ArH), 7.24 (1H, m, ArH), 7.11-7.00 (5H, m, ArH), 5.06 (1H, q, J = 2.1 Hz, ArH), 4.76 (1H, s, ArH), 4.40 (1H, d, J = 1.0 Hz, ArH), 3.63 (3H, s, ArH), 3.51 (3H, s, OCH3), 3.17 (1H, d, J = 2.2 Hz, ArH), 3.02 (1H, dd, J = 1.0, 0.9 Hz, ArH), 1.80 (3H, d, J = 2.1 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 15.7, 45.8, 48.1, 50.2, 54.1, 56.0, 57.4, 72.6, 106.3, 122.8,123.0, 123.4, 124.4, 124.5, 124.7, 124.9, 125.1, 125.6, 137.7, 138.5, 139.4, 142.1, 188.0, 202.7; HR-ESI MS calculated for C23H22O3 (M+H)+ 346.1641, found 346.1569. (10a); m.p. 275-277 ºC, 1H-NMR (300Hz, CDCl3) δ 7.88-7.85 (1H, m, ArH), 7.37-7.34 (1H, m, ArH), 7.32-7.28 (1H, m, ArH), 7.17-7.12 (5H, m, ArH), 4.87 (1H, s, ArH), 4.61 (1H, d, J = 3.1 Hz, ArH), 3.65 (1H, d, J = 8.6 Hz, ArH), 3.64 (3H, s, OCH3), 3.63 (3H, s, OCH3), 2.82 (1H, dd, J = 6.8, 3.2 Hz, ArH), 1.85 (3H, d, J = 6.3 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 17.2, 46.9, 47.8, 54.4, 55.8, 56.7, 58.7, 74.9, 107.6, 123.0, 123.7, 125.3, 125.6, 125.9, 126.1, 126.4, 126.8, 138.6, 139.5, 140.2, 143.6, 190.7, 204.2; HR-ESI MS calculated for C23H22O3 (M+H)+ 346.1641, found 346.1569.
13-Acetoxy-10-((S)-1-methoxyethyl)-11,12-dihydro-9H-9,10-[1,2]epicyclopenta-anthracen-15-one (10b)
To a solution of 15-Hydroxy-10-((S)-1-methoxymethyl)-11,12-dihydro-9H-9,10-[1,2]epi cyclopenta-anthracen-13-one (8c) 1.0 g (3.00 mmol, 1.0 equiv) in acetic anhydride 5 mL, iodide was added with stirring and then refluxed at 120ºC for 2 hours, The mixture was cooled to room temperature and quenched with water (10 mL) and stirred for a further 20 minutes, followed by extraction with CH2Cl2 (3×20 mL). The combined organic phase was dried over anh. Na2SO4, filtered and concentrated in vacuo. Purification of the residue by column chromatography (silica gel) with Hexane:EtOAc, (10:1) as the mobile phase gave 13-acetoxy-10-((S)-1-methoxyethyl)-11,12-dihydro-9H-9,10-[1,2]epicyclopenta-anthracen-15-one (10b) 1.0 g (90%); m.p. 215-217ºC; 1H-NMR (300Hz, CDCl3) δ 7.90 (1H, m, ArH), 7.37 (1H, m, ArH), 7.15 (6H, m, ArH), 5.75 (1H, s, ArH), 5.13 (1H, q, J = 6.3 Hz, ArH), 4.50 (1H, s, ArH), 3.73 (3H, s, ArH), 3.24 (2H, s, ArH), 2.32 (3H, s, OCH3), 1.85 (3H, d, J = 6.3 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 16.7, 21.4, 46.8, 49.8, 50.1, 55.1, 57.0, 73.5, 119.0, 123.8, 124.0, 124.5, 125.7, 125.9, 126.3, 126.5, 126.7, 138.4, 139.1, 140.3, 143.0, 165.8, 177.2, 204.9; HR-ESI MS calculated for C23H22O3 (M+H)+ 455.2018, found 455.1974.
General Procedure for the Grignard Addition to Cyclo-adduct 9,10
To a mixture of magnesium (5.0 equiv) in diethyl ether (5 mL) at 0ºC, alkyl/allyl bromide solution (1.0 equiv) was added and stirred for 30 minutes to give alkyl/allyl magnesium bromide solution as a turbid grey solution. To the cyclo-adduct 9,10 (0.20 mmol) in CH2Cl2 2 mL in a 50 ml round bottom flask at -78 ºC in an acetone/dry ice cooling bath, the solution of the Grignard reagent (0.40 mmol) was slowly added dropwise over 2 hours under argon. The resulting mixture was quenched with an aqueous saturated 20 mL NH4Cl solution and extracted with CH2Cl2 (3×20 mL). The combined organic phase was dried over anh. Na2SO4, filtered and concentrated in vacuo. Purification of the residue by column chromatography (silica gel) with Hexane:EtOAc, (10:1) as the mobile phase gave the desired Grignard addition products.
(9S,10S,11S,12S)-9-((S)-1-methoxyethyl)-15-methyl-10,11-dihydro-9H-9,10-[1,2]epicyclo pentaanthracen-13(12H)-one (11a)
Using the general procedure with the addition of MeMgBr (0.05 mL,0.40 mmol) to the cyclo-adduct 9 (0.07 g, 0.20 mmol), the title compound was obtained after column chromatography (0.055 g, 83%); m.p. 224-226ºC; 1H-NMR (300Hz, CDCl3) δ 7.86 (1H, m, ArH), 7.34 (1H, m, ArH), 7.18-7.05 (6H, m, ArH), 5.43 (1H, t, J = 1.2 Hz, ArH), 5.03 (1H, q, J = 6.3 Hz, ArH), 4.39 (1H, d, J = 3.0 Hz, ArH), 3.72 (3H, s, OCH3), 3.16 (1H, d, J = 6.3 Hz, ArH), 3.07 (1H, dd, J = 0.9, 4.8 Hz, ArH), 2.04 (3H, s, CH3), 1.82 (3H, d, J = 6.3 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 16.7, 17.1, 47.3, 51.4, 53.8, 55.3, 57.0, 73.4, 123.7, 124.0, 124.3, 125.6, 125.8, 126.0, 126.2, 126.6, 134.1, 138.5, 139.4, 140.5, 143.7, 176.7, 207.4; HR-EI MS calculated for C23H22O2 (M)+ 330.1630, found 330.1673.
9S,10S,11S,12S)-15-allyl-9-((S)-1-methoxyethyl)-10,11-dihydro-9H-9,10-[1,2]epicyclopen taanthracen-13(12H)-one (11b)
Using the general procedure with the addition of allylmagnesium bromide (0.07 mL, 0.60 mmol) to the cyclo-adduct 9 (0.07 g, 0.20 mmol), the title compound was obtained after column chromatography (0.053 g, 70%); m.p. 188-190ºC; ; 1H-NMR (300Hz, CDCl3) δ 7.85 (1H, m, ArH), 7.32 (1H, m, ArH), 7.12 (6H, m, ArH), 5.72 (1H, m, ArH), 5.45 (1H, d, J = 0.9 Hz, ArH), 5.15 (3H, m, ArH), 4.41 (1H, d, J = 2.1 Hz, ArH), 3.71 (3H, s, OCH3), 3.15 (3H, m, ArH, CH2), 3.07 (1H, d, J = 7.5 Hz, ArH), 1.81 (3H, d, J = 6.3 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 16.7, 29.7, 35.4, 47.4, 51.7, 52.4, 55.3, 57.0, 73.4, 123.4, 124.0, 124.4, 125.6, 125.8, 126.0, 126.2, 126.6, 132.5, 133.3, 138.6, 139.4, 140.5, 143.6, 178.4, 207.0; HR-EI MS calculated for C25H24O2 (M)+ 356.1776, found 356.1806.
(9S,10S,11S,12S)-9-((S)-1-methoxyethyl)-15-pentyl-10,11-dihydro-9H-9,10-[1,2]epicyclo pentaanthracen-13(12H)-one (11c)
Using the general procedure with the addition of pentanylmagnesium bromide (0.75 mL, 0.30 mmol) to the cyclo-adduct 9 (0.05 g, 0.15 mmol), the title compound was obtained after column chromatography (0.045 g, 78%); 1H-NMR (300Hz, CDCl3) δ 7.91 (1H, m, ArH), 7.38 (1H, m, ArH), 7.18 (6H, m, ArH), 5.48 (1H, d, J = 1.2 Hz, ArH), 5.19 (1H, q, J = 6.3 Hz, ArH), 4.42 (1H, d, J = 2.7 Hz, ArH), 3.76 (3H, s, OCH3), 3.18 (2H, m, ArH), 2.36 (2H, m, CH2), 1.85 (3H, d, J = 6.3 Hz, CH3), 1.53 (2H, m, CH2), 1.30 (4H, s, CH2), 0.93 (3H, t, J = 6.9 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 13.9, 16.7, 22.4, 26.2, 29.7, 31.0, 31.5, 47.6, 51.4, 52.7, 57.0, 73.4, 123.6, 123.9, 124.4, 125.5, 125.7, 125.9, 126.1, 126.6, 132.5, 138.6, 139.5, 140.6, 143.8, 181.1, 207.3; HR-EI MS calculated for C27H30O2 (M)+ 386.2246, found 386.2275.
(9S,10S,11S,12S)-15-hexadecyl-9-((S)-1-methoxyethyl)-10,11-dihydro-9H-9,10-[1,2]epi cyclopentaanthracen-13(12H)-one (11d)
Using the general procedure with the addition of hexadecylmagnesium bromide (2.00 mL, 6.40 mmol) to the cyclo-adduct 9 (1.00 g, 0.30 mmol), the title compound was obtained after column chromatography (0.12 g, 75%); m.p. 201-203ºC; 1H-NMR (300Hz, CDCl3) δ 7.86 (1H, m, ArH), 7.33 (1H, m, ArH), 7.11 (6H, m, ArH), 5.43 (1H, s, ArH), 5.15 (1H, q, J = 6.3 Hz, ArH), 4.38 (1H, d, J = 6.3 Hz, ArH), 3.72 (3H, s, OCH3), 3.15 (1H, d, J = 6.3 Hz, ArH), 3.13 (1H, d, J = 6.6 Hz, CH2), 2.35 (2H, m, CH2), 1.82 (3H, d, J = 6.3 Hz, CH3), 1.44 (2H, m, CH2), 1.26 (26H, s, CH2), 0.88 (3H, t, J = 6.3 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 13.1, 15.6, 21.7, 22.9, 25.5, 25.9, 28.4, 28.5, 28.7, 29.3, 30.0, 30.9, 40.9, 46.6, 50.4, 51.6, 54.3, 56.0, 72.4, 122.6, 122.9, 123.4, 124.5, 124.7, 124.9, 125.1, 125.6, 131.4, 137.6, 138.4, 139.6, 142.7, 180.0, 206.3; HR-EI MS calculated for C38H52O2 (M)+ 540.3967, found 540.4010.
(9S,10S,11S,12R)-9-((S)-1-methoxyethyl)-13-methyl-10,11-dihydro-9H-9,10-[1,2]epicyclopentaanthracen-15(12H)-one (12a)
Using the general procedure with the addition of methylmagnesium bromide (0.07 mL, 0.60 mmol) to the cyclo-adduct 10 (1.00 g, 0.30 mmol), the title compound was obtained after column chromatography (0.07, 70%); m.p. 232-234ºC; 1H-NMR (300Hz, CDCl3) δ 7.85 (1H, m, ArH), 7.21 (7H, m, ArH), 5.59 (1H, s, ArH), 4.79 (1H, q, J = 6.0 Hz, ArH), 4.58 (1H, d, J = 3.0 Hz, ArH), 3.68 (3H, s, OCH3), 3.65 (1H, d, J = 6.9 Hz, ArH), 2.76 (1H, dd, J = 3.0, 6.0 Hz, ArH), 2.15 (3H, s, CH3), 1.86 (3H, d, , J = 6.3 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 17.1, 19.9, 47.0, 52.5, 55.5, 55.7, 74.9, 123.0, 123.6, 125.1, 125.5, 126.0, 126.1, 126.4, 126.6, 135.7, 136.8, 138.4, 139.5, 140.9, 143.7, 178.5, 207.8; HR-EI MS calculated for C23H22O2 (M)+ 330.1630, found 330.1673.
Results and Discussion
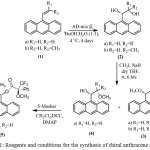 |
Scheme 1: Reagents and conditions for the synthesis of chiral anthracene auxiliary |
The synthesis of the chiral anthracene auxiliary started with vinyl anthracenes (1a-b) which was prepared according to the literature.14 Asymmetric dihydroxylation with AD-mix β was prepared using literature procedure16 to afford compounds 2a-b in fair yields. Then methylation of 2a with CH3I and NaH in THF for 6 hours at room temperature gave the dimethoxy compound 3a in 52% yield and the mono-methylated product 4a in 36% yield. Methylation of 2b using the same conditions gave only the dimethylated product 3b in 18% yield. This low yield might be due to the steric strain of the methyl side chain of the propyl group. However, the methylated product yield was improved when the reaction time was increased.
The absolute stereochemistry of 2a was confirmed by comparing the spectroscopic data and optical rotation of the previous report.17 To determine enantiomeric purity of 2a, (R)-9-(1,2-dimethoxyethyl)anthracene 4a was treated wtih (–)-(S)-a-Methoxy-a-(trifluoromethyl)phenylacetic acid [(-)-(S)-MTPA] in the presence of DCC and DMAP. These reaction afforded (-)-(S)-MTPA ester 5a as a single diastereomer (dr ≥ 20:1) after structural elucidation. The low yield of 5a might be due to the use of (-)-(S)-MTPA-acid reacted with steric alcohol 4a. The anthracene 6 was synthesised according to Snyder’s method and the structure confirmation was compared to the literature.15
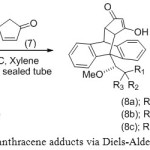 |
Scheme 2: Enolic anthracene adducts via Diels-Alder reactions |
The Diels-Alder reactions of the chiral anthracene 3a-b and 6 with cyclopentene-3,5-dione (7) refluxing in xylenes for 12 hours resulted in the ketone forms of 8a- c as single diastereomers (Scheme 2). The structures of the addition adducts were confirmed by NMR spectroscopy. The 13C NMR spectra appeared to have peaks around 203 and 211 ppm which suggested the solution structures to be diketone carbonyl groups of 8a-c. However, the single crystal X-ray crystallography of adduct 8c were showed to be an enolic anthracene adduct (Figure 2). These were supported the previous study19 that cyclopentene-3,5-dione itself in refluxing benezene was present exclusively as the keto tautomer. The X-ray crystallography also showed the orientation of the methoxy group away from the approaching dienophile. These observations led to a proposition that in the transition state, the facial selectivity is controlled by minimisation of electrostatic repulsion between the methoxyl oxygen and the approaching dienophile. While the cyclo-addition completed, hydrogen bonding helped to stabilise the alternative product, giving rise to a single diastereomer as depicted in Scheme 2.
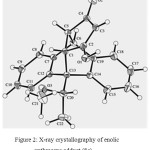 |
Figure 2: X-ray crystallography of enolic anthracene adduct (8c) |
Prior to transformation of the carboxyl group of 8c, the hydroxyl group was converted to either an ether or ester derivative with good to poor regio-selectivity depending on the nature of the protection employed (Table 1). Transformation of 8c into enolate anions led to the breaking of the hydrogen bond, and consequently delocalisation of the enolate anion gave two regio-isomers (Scheme 3). The regio-selectivity obtained was due to the steric hindrance of substituents. The small steric group of the –CH3 gave the methyl ether low selectivity and about a 2:3 ratio of adducts (9) and (10), respectively. Meanwhile, the -Ac group had high selectivity and gave exclusively adduct (10) (Table 1, entry 2). The HMBC analyses were used in assigning the regio-chemistry of the ether/ester adduct. In the regio-isomers (10), the HMBC spectra showed correlations between ether/ester carbons and proton at C2².
Table 1: Condition and reagent of the synthetic adducts (9) and (10)
|
Entry |
Conditions |
R |
% Yield |
|
1 |
NaH, CH3I, DMF |
Me |
9a: 10a = 30: 50 |
|
2 |
Ac2O, I2 |
Ac |
9b: 10b = 0: 90 |
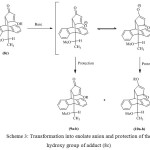 |
Scheme 3: Transformation into enolate anion and protection of the hydroxy group of adduct (8c) |
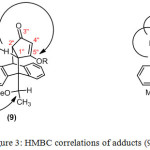 |
Figure 3: HMBC correlations of adducts (9) and (10) Click here to View figure |
Treatment of enol ethers (9) and (10) with Grignard reagents gave 1,2-addition and then hydrolysis of the resulted products gave the corresponding β-alkylketone anthracene adducts (11) and (12), respectively (Scheme 4 and 5). The organomagnesium compounds added to the carbonyl group subsequently caused hydrolytic cleavage of the enol ether and elimination of water16 to give 11 and 12. In this approach, steric hindrance plays an important role as using bulky Grignard reagents gave only product (11) from the addition to the less hindered carbonyl ketone (9). While the hindered carbonyl ketone 10 was not attacked by the bulky Grignard reagents (Table 2). The 1H NMR spectra of (11) indicated the absence of the methoxy protons and the presence of the alkyl protons in high fields and the 13C NMR spectra showed the present of carbonyl group at around δ 207.4 ppm. The HMBC spectra showed the correlations between C-H proton of the alkyl groups and the C1², C2² and C4², correlations between H4² proton and the C5 carbonyl group, and correlations between H1² proton and C9 anthracene substituent. The COSY spectra showed the correlations between H1² and H2² and the long length coupling between C-H proton of the alkyl groups and H2² and H4² protons. Thus, these NMR experiments assigned the carbonyl group to be on the same side as the C9 anthracene substituent. The COSY spectra of 12 showed the long length coupling between H1² and C-H proton of the alkyl group, but correlation between H2² and C-H proton of the alkyl group was not observed.
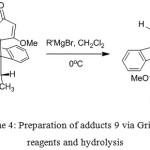 |
Scheme 4: Preparation of adducts 9 via Grignard reagents and hydrolysis |
Table 2: Results of the synthesis adducts 11a-d
|
Entry |
Product |
R |
Yields (%) |
|
1 |
11a |
CH3 |
83 |
|
2 |
11b |
CH2CH=CH2 |
70 |
|
3 |
11c |
C5H11 |
78 |
|
4 |
11d |
C16H33 |
75 |
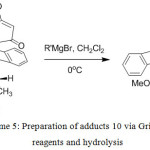 |
Scheme 5: Preparation of adducts 10 via Grignard reagents and hydrolysis |
Table 3: Results of the synthesis adducts 12a-d
| Entry | 10 |
Product |
R | Yields (%) |
| 1 |
R= CH3 |
12a |
CH3 | 70 |
| 2 |
12b |
CH2CH=CH2 | – | |
| 3 |
12c |
C5H11 | – | |
| 4 |
12d |
C16H33 | – | |
| 5 | R= Ac |
12a |
CH3 | – |
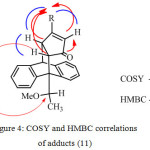 |
Figure 4: COSY and HMBC correlations of adducts (11) Click here to View figure |
Conclusions
Chiral anthracene templates were synthesised prior to use in Diels-Alder reactions with cyclopentene-3,5-dione. The results showed that the cyclo-adducts were obtained with good regio-selectivity as a single diastereomer from completely enolic forms in crystal structure and diketone forms in the solution structures. However, the studies on the stereo-selectivity using organomagnesium addition to the methoxyenones resulted in the cleavage of enol ether and elimination of water. These could undergo hydrogenation of the enone double bond to give a chiral cyclopentenones . On the other hand, studies with other nucleophilic additions to the carbonyl group without loss of stereo-centre should be investigated.
Acknowledgement
We thank the Department of Chemistry, Faculty of Science, Silpakorn University for the financial support for Y. J. We are highly grateful to Dr. Anthony C. Willis, Research School of Chemistry, Australian National University, Australia, for help in the studies of X-ray data. We also thank The Chulabhorn Research Institute for HRMS spectroscopy.
References
- Kikuchi, H.; Tsukitani, Y.; Iguchi, K.; Yamada, Y. Tetrahedron Letters 1983, 24,1549.
CrossRef - Ishibashi, M.; Takeuchi, S.; Kobayashi, J. Tetrahedron Lett., 1993, 34, 3749.
CrossRef - Sugawara, F.; Kuramochi, K.; Saito, F.; Takeuchi, R.; Era, T.; Takemura, M.; Kobayashi, J.; Sakagushi, K.; Kobayashi, S. Tetrahedron 2006, 62, 8006-8015.
CrossRef - Fukushima, S.; Takeuchi, Y.; Kishimoto, S.; Yamashita, S.; Uetsuki, K.; Shirakawa, S.; Suzuki, M.; Furuta, K.; Noyori, R.; Sasaki, H.; Kikuchi, Y.; Kita, T.; Yamori, T.; Sawada, J.; Kojima, M.; Hazato, A.; Kurozumi, S.; Fukushima, M. Anti-cancer drugs 2001, 12,221.
CrossRef - (a) Blumenkopf, T.A.; Overman, L.E. Chem. Rev., 1986, 86,857. (b) Habermas, K.L.; Denmark, S.E.; Jone, T.K. Org. React. 1994, 45, 1.
CrossRef - (a) Brummond, K.M.; Kent, J.L. Tetrahedron, 2000, 56, 3263. (b) Perez-serrano, L.; Blanco, Urgoiti, J.; Cararrubios, L.; Dominguez, G.; Rerez-Castells, J. J. Org. Chem. 2000, 65, 3513.
CrossRef - Vavie, C.P.; Danheiser, R.L. Angew Chem Int. Ed. Engl. 2005, 12, 44, 5867.
- Thebtaranonth, Y. Pure & Appl. Chem. 1997, 69, 609 and references there in.
- Burrgess, K.L.; Corbett, M.S.; Eugenio, P.; Lajkiewicz, N.J.; Liu, X.; Sanyal, A.; Yan, W.; Yuan, Q.; Snyder, J.K. Bioorg. Med. Chem. 2005, 13, 5299.
CrossRef - Adams, H.; Jones, S.; Meijer, A.J.H.M.; Najah, Z.; Ojea-Jimenez, I.; Reeder, A.T. Tetrahedron : Asymmetry, 2011, 22,1620.
CrossRef - Jones, A.L.; Liu, X.; Snyder, J.K. Tetrahedron Lett. 2010, 51, 1091.
CrossRef - Adams, H.; Jones, S.; Meijer, A.J.H.M.; Najah, Z.; Ojea-jimenez, I.; Reeder, A.T. Tetrahedron : Asymmetry, 2011, 22,1620.
CrossRef - Depuy, C.H.; Lyons, C.E. J.Am. Chem. Soc. 1960, 82, 631.
CrossRef - Hawkins, E. G. E. J. Chem. Soc. 1957, 3858-3862.
CrossRef - Sanyal, A.; Snyder, J. K. Org. Lett. 2000, 2, 2527.
CrossRef - Corey, E.J.; Noe, M.C. J. Am. Chem. Soc. 1996, 118, 11038-11053.
CrossRef - Snyder, J.K.; Kerrie, L.B.; Neil, J.L.; Amitav, S.; Wanlin, Y. Org. Lett, 2005, 7, 31-34.
CrossRef - Hall, D.G.; Rauniyar, V.; Zhai, H. J. Am. Chem. Soc. 2008, 130, 8481-8490.
CrossRef - Giorgi, G.; Lampariello, L. R.; Minetto, G.; Paoli, M.L.; Riello, V.; Rodriquez, M.; Sega, A. Eur. J. Org. Chem. 2003, 4777.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


