Design of Cinnamic Acid Derivatives and Molecular Docking Toward Human-Neutral α-Glucosidase by Using Homology Modeling
Teni Ernawati1,2, Abdul Mun’im1, Muhammad Hanafi2 and Arry Yanuar1
1Faculty of Pharmacy – Universitas Indonesia, Depok 16242, Indonesia.
2Research Center for Chemistry – Indonesian Institute of Sciences (LIPI) Kawasan Puspiptek, Serpong Tangerang Selatan, banten 15314.
Corresponding Author E-mail: arry.yanuar@ui.co.id
DOI : http://dx.doi.org/10.13005/ojc/330512
Most of the biological testing of α-glucosidase inhibitors was carried out on yeast and rat intestinal. The aim of this study was to explore molecular interactions which occurred during inhibition process of human-neutral α-glucosidase by cinnamic acid derivatives. In this paper, cinnamic acid was used as the lead compound whose carboxylic acid moeity was replaced by alkyl amine against the macromolecule target which was human-neutral α-glucosidase enzyme. In order to understand the mechanism of interaction of the ligand binding conformations and to identify potent α-glucosidase inhibitor, molecular modeling studies with homology modeling method were used to investigate the problem. The structure of homology model of human α-glucosidase was built by using neutral-human α-glucosidase (GANC) with 914 amino acid residues from the Swiss-Prot with Q8TET4 identity and using the mold template of 2G3M (PDB ID). The model of enzyme was constructed based on the crystal structure of the S.solphataricus α-glucosidase, Ma1A, and human N-terminal subunit of Maltase Glucoamylase (NtMGAM). The molecular docking simulation of cinnamic acid derivatives was carried out on Autodock 4.2. Two parameters from docking simulation that can predict the inhibition activity of α-glucosidase enzyme are: low free energy Gibs and low kinetic inhibition value. The Gibs free energy value (ΔG) obtained from the docking simulation showed that 13 cinnamic acid derivatives compounds had an affinity for the receptor α-glucosidase. These compound could act as α-glucosidase inhibitor.
KEYWORDS:Docking; α-glucosidase Inhibitors; Cinnamic Acid Derivatives; Neutral-Human α-Glucosidase; Virtual Screening
Download this article as:| Copy the following to cite this article: Ernawati T, Mun’im A, Hanafi M, Yanuar A. Design of Cinnamic Acid Derivatives and Molecular Docking Toward Human-Neutral α-Glucosidase by Using Homology Modeling. Orient J Chem 2017;33(5). |
| Copy the following to cite this URL: Ernawati T, Mun’im A, Hanafi M, Yanuar A. Design of Cinnamic Acid Derivatives and Molecular Docking Toward Human-Neutral α-Glucosidase by Using Homology Modeling. Orient J Chem 2017;33(5). Available from: http://www.orientjchem.org/?p=37593 |
Introduction
α-Glucosidase enzymes (EC 3.2.1.20) are classified into GH13 and GH31 classes.1 These enzymes are the kind of exo-glycosidase that hydrolyzes α-D-glucopyranoside. During the hydrolysis of α-glucosidase, GH13 and GH31 α-glucosidase enzymes generate α-D-glucopyranose with retention of the configuration of the anomeric position.2 Inhibition of the α-glucosidase enzyme make glucose levels in the blood return within normal range.3 Glucosidase is responsible for the catalytic cracking glycosidic bonds specifically depending on the number of monosaccharides, the position of the cleavage site, and the configuration of the hydroxyl group in the substrate.4 The main mechanism of enzymatic hydrolysis of glycosidic bonds proposed by Koshland is explained through the mechanism of detention and inversion in anomeric configuration.5 Compounds that can inhibit α-glucosidase enzyme called α-glucosidase inhibitors (AGIs). AGIs compounds are widely used for the treatment of type 2 diabetes patients.6-8 These inhibitors play a vital role in glycemic control. α-Glucosidase inhibitor compound inhibits α-glucosidase enzyme contained in the intestinal wall. Diabetes drugs functioning as α-glucosidase inhibitors are acarbose, miglitol, and voglibose.3,9-11 The α-glucosidase inhibitor is recommended as first-line therapy by the International Diabetes Federation (IDF) and the American Association of Clinical Endocrinologists (AACE). AGI’s efficacy, safety, tolerability cardiovascular benefits, and lack of hypoglycemia make them suitable for diabetics 12.
Research in the field of organic chemistry of the cinnamic acid derivatives has also been growing. In the last decades, a lot of research and development of the cinnamic acid derivatives as α-glucosidase inhibitor has been done. Some cinnamic acid derivatives which have inhibitory activity against α-glucosidase; such as aegeline, aegelinoside A and B, phenylethylcinnamide, aloeresin A, flavonol glycosides cinnamate.13 Several studies of structure activity relationship of α-glucosidase inhibitor class of cinnamic acid derivatives indicated that the substituent hydroxy or methoxy at position 4 in the trans-cinnamic acid could increase the activity of inhibitory of α-glucosidase enzyme. Likewise, the addition of ethylester group on the carboxylic group of cinnamic acid plays an important role to the inhibition of α-glucosidase 14.
Based on the results of the literature study, it was not yet known whether the cinnamic acid compounds whose carboxylic acid moeity was replaced by alkyl amine had good activity on α-glucosidase inhibitor or not. In this study, we analyzed 50 designs of cinnamic acid derivatives using molecular docking simulation that involves homology modeling and ligand-based virtual screening. Molecular docking remained an important tool for structure ligand based screening to find new active ligand. Molecular docking was widely used to predict protein-ligand complexes and to screen several compounds that would arrange the activity of a bi-logical receptor.15 Ligand-based virtual screening approach was used in here to calculate the lowest binding energy between α-glucosidase enzyme with ligand and also to study their binding affinity 16. We chose the enzyme α-glucosidase C-neutral protein in humans as the target for virtual screening.17-24
Materials and Method
Homology Modeling
The target protein to be used was α-glucosidase derived from human neutral α-glucosidase. This protein was not yet available at the provider’s site Protein Data Bank (PDB) so we were looking for a target protein in Gene Bank sequence database which could be accessed at http://www.ncbi.nlm.nih.gov/genbank/. This database was created by the National Center for Biotechnology Information (NCBI). We chose the corresponding protein to be made using Swiss-Model homology with existing mold in the Protein Data Bank (PDB) which could be accessed at www.pdb.org. For the target protein we chose UniprotKB/Swiss-Prot: Q8TET4 then it was made as homology model with existing mold on PDB ID 2G3M. Model templates made by Swiss-Model had a similarity sequence identity of 31.35%.
Validation
Validation was done by comparing the value of the bond between the active ligand- receptor ligand binding site that comparison receptor ligand binding site. Analysis of the data was revealed by RMSD (Rate Mean Square Deviation) value ratio. Docking method is said to be good if its RMSD value is less than or equal to 2.0 (≤ 2.0). Macromolecules that have been superposed were prepared for molecular docking. Macromolecules were optimized by using software Autodock 4.2. 3D structure of macromolecules added hydrogen atoms was then repaired of the charge by adding the partial charge Gasteiger and given force field using Autodock. Macromolecular structure and ligand to be docked firstly were optimized then after that each macromolecules and ligand were made their PDBQT file, also grid file parameter file (GPF), and docking file parameter file (DPF). GPF would inform Autogrid which potential receptors needed to be calculated and the type of map types that must be counted and their location. While the DPF would inform Autodock which folder to be used, which ligand to be moved, including the center and the torsion of the ligand, what docking algorithm to be used, and the number of docking should be done.
Visualization
The results of molecular docking were visualized by using Autodock and Chimera. Docking outcome parameters were analyzed. Receptor-ligand binding energy obtained from the docking was sorted by their lowest energy. And through Chimera, it was observed that amino acid residue is the closest to the ligand.
Materials
The molecular docking studies were carried out to understand the binding interaction modes of α-glucosidase enzyme with α-glucosidase inhibitors. The structures of known α-glucosidase inhibitors such as acarbose, 1-deoxynojirimycin, miglitol and voglibose were used as references, shown in Table 1. The structures of cinnamic acid derivatives compounds already have the IC50 data, shown in Table 2. The structures of cinnamic acid derivatives that were to be tested are shown in Table 3.
Result and Discussion
Most of the α-glucosidase inhibitors testing were performed on yeast and rat intestinal. However, 3D protein structure of α-glucosidase unfortunately so far was not available hence the structure homology modeling for α-glucosidase were still being developed. In this paper, we observed molecular interactions of human α-glucosidase when it was inhibited by cinnamide derivatives using lead compound of cinnamic acid. The model of the enzyme was constructed based on the crystal structure of the S.solphataricus α-glucosidase, Ma1A, and human N-terminal subunit of Maltase Glucoamylase (NtMGAM). Moreover, the model were also including homology modeling of human α-glucosidase (GANC) with 914 amino acid residues from the Swiss-Prot Q8TET4 identity and using the template mold 2G3M (PDB ID). 2G3M structure is a crystal structure of α-glucosidase from Sulfolobus solfataricus Maia which was taken as a template for the Swiss-Model showed the similarity of 31.35%. Ramachandaran plot of the results of the model obtained showed the following results. The residues on the most favored regions have 391 residues with a percentage of 88.5%. Meanwhile, the residues in additional allowed regions have as much as 46 residues with percentage of 10.4%. In addition, the residues in generously allowed regions have as much as 2 residues with percentage of 0.5%. Finally, the residues in disallowed regions have as much as 3 residues with percentage of 0.7%.
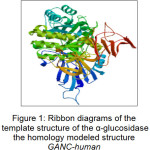 |
Figure 1: Ribbon diagrams of the template structure of the α-glucosidase the homology modeled structure GANC-human Click here to View figure |
In this paper, the co-crystal ligand was detached from the active site and re-docked using AutoDock 4.2 into the binding pocket to calculate the rmsd value. The rmsd value was found to be 1.458 Å that showed the extent of overlapping of docked pose and actual pose of the co-crystal ligand. Ribbon diagrams of the template of the homology modeled structure of GANC-human α-glucosidase are shown in Figure 1.
The molecular docking studies were used to predict the interactions between cinnamic acid derivatives with the human C-neutral (GANC) α-glucosidase enzyme. Herein, we used reference compound as the standard. Results of the analysis indicated by the value of molecular docking Gibs free energy (ΔG) was shown in the table 1 below:
Table 1: Docking Scores of The known α-glucosidase inhibitor
| No. | Reference sample | Result of Docking | IC50 (mM) | |
| -∆G (kcal/mol) | Ki (µM) | |||
| 1 | Acarbose (1) | -2.88 | 7.77 | 199.81 |
| 2 | 1-deoxynojirimycin (2) | -6.07 | 35.53 | 5.60 |
| 3 | Miglitol (3) | -5.04 | 201.15 | 14.47 |
| 4 | Voglibose (4) | -3.55 | 2.50 | 276.86 |
Parameters measured were free energy Gibs (-ΔG in kcal/mol) which were resulted from receptor-ligand interactions. Table 1 showed the result of docking simulation in column 2 and 3 and results from in vitro testing of references samples in column 4. It can be seen that there is significant corelation between docking simulation results and in vitro testing results. Table 1 also showed that 1-deoxynojirimycin capable of inhibiting the human-neutral α-glucosidase enzyme and also was ranked as the highest among all the known α-glucosidase inhibitors. For reference compounds such as miglitol (3), voglibose (4) and acarbose (1), they also have the ability to inhibit the human-neutral α-glucosidase enzyme but it is still below the inhibition levels of 1-deoxynojirimycin because they have larger value Gibs free energy (ΔG) than that of 1-deoxynojirimycin. Their Gibs free energies (ΔG) were -5.04 kcal/mol, -3.55 kcal/mol, and -2.88 kcal/mol respectively.
The best ranked for the known α-glucosidase inhibitor was shown in Figure 2. This figure showed that 1-deoxynojirimicyn (1) compound was reported to form hydrogen bonds (shown as a green breaking point) with the amino acid residues HIS645, ASP398, ASP 511, ASP587, and ARG571.
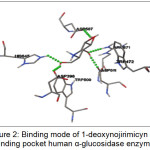 |
Figure 2: Binding mode of 1-deoxynojirimicyn in binding pocket human α-glucosidase enzyme |
The simulation analysis results of molecular docking of the tested compounds are shown in the Table 2. 20 compounds which were already known of their in-vitro activities against α-glucosidase enzyme are simulated in molecular docking software in order to compare the results from in-vitro testing. We could estimate that the extent to which the activeness of the sample compounds of docking results and the predictability of its activeness in inhibiting human α-glucosidase enzyme. The results of the docking simulation of 20 tested compounds that have Gibs free energy values (ΔG) better than that of 1-deoxynojirimicyn are compounds of 21, 22 and 24 with a value of ΔG = -6.11 kcal/mol, -6.67 kcal/mol, and -6.76 kcal/mol respectively. It can be seen that there is significant correlation between docking simulation results (which have low Gibs free energy value and low kinetic inhibition value) and in vitro testing results.
Table 2: Docking Scores of Tested Compound against human α-glucosidase enzyme
| No | Tested Samples | R1 | R2 | R3 | R4 | R5 | Log P | IC50 | ∆G (kcal/mol) | Ki(µM) |
| 1 | Compound 5 | H | H | H | H | OH | 1.93 | > 5 mM | -3.42 | 3.12 |
| 2 | Compound 6 | H | H | H | H | OC2H5 | 2.95 | > 5 mM | -3.79 | 1.67 |
| 3 | Compound 7 | H | H | H | OH | OH | 1.54 | > 5 mM | -3.80 | 1.65 |
| 4 | Compound 8 | H | H | OH | H | OH | 1.54 | 1.27 mM | -3.83 | 1.57 |
| 5 | Compound 9 | H | OH | H | H | OH | 1.54 | 0.20 mM | -3.47 | 2.86 |
| 6 | Compound 10 | H | H | H | OCH3 | OH | 1.81 | 4.34 mM | -3.59 | 2.35 |
| 7 | Compound 11 | H | H | OCH3 | H | OH | 1.81 | 0.58 mM | -3.70 | 1.93 |
| 8 | Compound 12 | H | OCH3 | H | H | OH | 1.81 | 0.04 mM | -3.26 | 4.06 |
| 9 | Compound 13 | H | OCH3 | H | H | OC2H5 | 2.41 | 0.05 mM | -3.74 | 1.82 |
| 10 | Compound 14 | H | NO2 | H | H | OH | 1.92 | > 5 mM | -4.17 | 876.64 |
| 11 | Compound 15 | H | OC6H13 | H | H | H | 3.88 | > 5 mM | -4.56 | 453.13 |
| 12 | Compound 16 | H | OC4H9 | H | H | OH | 3.05 | > 5 mM | -4.11 | 973.62 |
| 13 | Compound 17 | H | F | H | H | OH | 2.09 | 0.27 mM | -3.43 | 3.07 |
| 14 | Compound 18 | H | Cl | H | H | OH | 2.49 | 0.39 mM | -4.30 | 702.4 |
| 15 | Compound 19 | H | Br | H | H | OH | 2.76 | > 5 mM | -3.66 | 2.09 |
| 16 | Compound 20 | H | OPhe | H | H | OH | 3.47 | 0.44 mM | -5.19 | 157.79 |
| 17 | Compound 21 | H | OBn | H | H | OH | 3.54 | > 5 mM | -6.11 | 3.72 |
| 18 | Compound 22 | H | H | H | H | NHCHCHPhe | 3.66 | 0.0358 mM | -6.67 | 12.96 |
| 19 | Compound 23 | H | OH | H | H | Phe | 3.2 | 0.0410 mM | -5.21 | 151.11 |
|
20 |
Compound 24 | OH | OH | H | H | Phe | 2.81 | 0.0621 mM | -6.76 |
11.06 |
The results for the simulation of the 20 tested compounds are shown in Fig.3. Compound 22 was reported to form hydrogen bonds, shown as a green breaking point, with the amino acid residues ASP537 while the compound 24 was reported to form hydrogen bonds, shown as a green breaking point, with the amino acid residues ASP (398, 511) and TRP472.
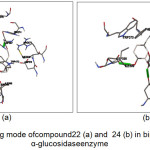 |
Figure 3: Binding mode of compound 22 (a) and 24 (b) in binding human α-glucosidase enzyme |
Table 3 showed the results of docking simulation of 50 cinnamic acid derivatives which were molecular docked against C-neutral-human α-glucosidase enzyme. There were as many as 13 compounds (compounds of 27, 28, 37, 51, 52, 55, 58, 59, 64, 65, 66, 67 and 68) that have free bond energy value (ΔG) better than that of 1-deoxynojirimycin compound. It can be seen that there is significant correlation between docking simulation results (which have low Gibs free energy value and low kinetic inhibition value) and in vitro testing results. This means that these 13 cinnamic acid derivative compounds would tend to bind to the receptor C-humans α-glucosidase than the other cinnamic acid derivatives. Gibs free energy value (ΔG) of the docking results showed that 13 cinnamic acid derivatives have affinities for the receptor of C-Neutral human α-glucosidase that could act as α-glucosidase inhibitor.
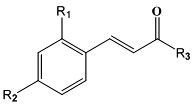
Table 3: Docking Scores of sample compounds ligand against human α-glucosidase enzyme
| No | Sample compounds | R1 | R2 | R3 | log P | ∆G (kcal/mol) | Ki (µM) |
| 1 | Compound 25 | H | NH2 | OH | 1.13 | -2.15 | 26.7 |
| 2 | Compound 26 | NH2 | H | OH | 1.13 | -2.52 | 14.12 |
| 3 | Compound 27 | H | NH2 | NH2 | 0.48 | -6.85 | 9.55 |
| 4 | Compound 28 | H | OCH3 | NH2 | 1.16 | -6.60 | 14.58 |
| 5 | Compound 29 | H | OCH3 | NHCH3 | 1.39 | -4.07 | 1.04 |
| 6 | Compound 30 | H | OCH3 | NHC2H5 | 1.73 | -4.30 | 703.98 |
| 7 | Compound 31 | H | OCH3 | NHC3H7 | 2.22 | -4.56 | 452.27 |
| 8 | Compound 32 | H | OCH3 | NHCH(CH3)2 | 2.05 | -4.62 | 408.15 |
| 9 | Compound 33 | H | OCH3 | NHC8H17 | 2.05 | -5.71 | 65.77 |
| 10 | Compound 34 | H | OCH3 | NH-Phe | 3.05 | -6.03 | 37.81 |
| 11 | Compound 35 | H | OH | NH2 | 0.89 | -3.39 | 3.29 |
| 12 | Compound 36 | H | OH | NHCH3 | 1.13 | -4.15 | 914.98 |
| 13 | Compound 37 | OH | H | NHCH3 | 1.13 | -6.13 | 32.32 |
| 14 | Compound 38 | H | OH | NHC2H5 | 1.47 | -4.31 | 697.19 |
| 15 | Compound 39 | H | OH | NHC3H7 | 1.95 | -4.52 | 488.08 |
| 16 | Compound 40 | H | OH | NHC8H17 | 4.04 | -5.86 | 50.36 |
| 17 | Compound 41 | H | NH2 | NHCH3 | 0.71 | -5.43 | 105.05 |
| 18 | Compound 42 | H | NH2 | NHC2H5 | 1.05 | -5.76 | 59.82 |
| 19 | Compound 43 | H | NH2 | NHC8H17 | 3.63 | -5.91 | 46.31 |
| 20 | Compound 44 | H | OH | NH-Phe | 2.79 | -5.00 | 216.62 |
| 21 | Compound 45 | H | NH2 | NH-Phe | 2.38 | -3.58 | 2.39 |
| 22 | Compound 46 | H | OCH3 | NH-Phe-OCH3 | 2.93 | -5.02 | 207.54 |
| 23 | Compound 47 | H | NH2 | NH-Phe-OCH3 | 2.25 | -5.98 | 41.14 |
| 24 | Compound 48 | H | OCH3 | NH-Glu | -0.25 | -5.71 | 65.37 |
| 25 | Compound 49 | H | NH2 | NH-Glu | -0.93 | -5.24 | 143.35 |
| 26 | Compound 50 | H | OCH3 | O-Glu | 0.43 | -5.66 | 71.17 |
| 27 | Compound 51 | H | NH2 | O-Glu | -0.25 | -6.22 | 27.65 |
| 28 | Compound 52 | NO2 | H | OH | 1.92 | -7.06 | 6.71 |
| 29 | Compound 53 | H | NO2 | OH | 1.92 | -4.15 | 866.02 |
| 30 | Compound 54 | NO2 | NO2 | NH2 | 1.28 | -4.93 | 241.40 |
| 31 | Compound 55 | NO2 | H | NH2 | 1.28 | -7.78 | 1.97 |
| 32 | Compound 56 | H | NO2 | NHCH3 | 1.71 | -5.93 | 45.32 |
| 33 | Compound 57 | H | NO2 | NHC2H5 | 1.92 | -6.05 | 36.74 |
| 34 | Compound 58 | H | NO2 | NHC3H7 | 2.37 | -6.10 | 33.99 |
| 35 | Compound 59 | H | NO2 | NHCH(CH3)2 | 2.24 | -6.17 | 30.24 |
| 36 | Compound 60 | H | NO2 | NHC8H17 | 2.24 | -5.70 | 65.87 |
| 37 | Compound 61 | H | NO2 | NH-Phe | 3.27 | -5.47 | 97.29 |
| 38 | Compound 62 | H | H | NHCH3 | 1.52 | -4.07 | 1.04 |
| 39 | Compound 63 | NO2 | H | NHCH3 | 1.71 | -5.88 | 48.83 |
| 40 | Compound 64 | NO2 | H | NHC2H5 | 1.92 | -6.40 | 20.48 |
| 41 | Compound 65 | NO2 | H | NHC3H7 | 2.37 | -6.12 | 32.93 |
| 42 | Compound 66 | NO2 | H | NHC8H17 | 4.65 | -6.67 | 12.87 |
| 43 | Compound 67 | NO2 | H | NH-Phe | 3.27 | -7.25 | 4.85 |
| 44 | Compound 68 | NO2 | H | NH-Phe-OCH3 | 3.40 | -6.63 | 13.78 |
| 45 | Compound 69 | H | NO2 | NH-Phe-OCH3 | 3.40 | -5.58 | 81.73 |
| 46 | Compound 70 | H | H | NHC2H5 | 1.86 | -4.27 | 738.43 |
| 47 | Compound 71 | H | H | NHC3H7 | 2.34 | -4.49 | 513.16 |
| 48 | Compound 72 | H | H | NHCH(CH3)2 | 2.17 | -4.62 | 411.26 |
| 49 | Compound 73 | H | H | NHC8H17 | 4.43 | -5.73 | 62.67 |
| 50 | Compound 74 | H | H | NH-Phe | 3.18 | -5.27 | 136.67 |
The best compounds from the docking simulation are shown in Fig.4. Compound 27 was reported to form hydrogen bonds, shown as a green breaking point, with the amino acid residues ASP587 and ARG643. Compound 52 was reported to form hydrogen bonds, shown as a green breaking point, with the amino acid residues TRP584 and ARG571. Compound 55 was reported to form hydrogen bonds, shown as a green breaking point, with the amino acid residues ASP (587, 616). Compound 67 were reported to form hydrogen bonds, shown as a green breaking point, with the amino acid residues ASP (398, 587).
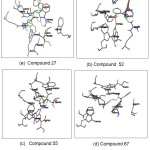 |
Figure 4: Binding mode of compounds 27(a), 52(b), 55(c) and 67(d) in binding pocket human α-glucosidase enzyme Click here to View figure |
Conclusion
In this paper, the molecular studies were conducted on 50 cinnamic acid derivatives against C-Neutral human α-glucosidase enzyme. The docking analysis resulted in the ligand interactions with the binding site. The binding interaction of the ligands to the enzyme indicated that conserved amino acidw residues in GANC-human protein played an important role in maintaining a functional conformation and were involved in binding to the inhibitors. The interactions mechanism of the protein and inhibitors in this study were useful to understand the potential mechanism of the protein of GANC-human and α-glucosidase inhibitors interactions. Two parameters from docking simulation that can predict the inhibition activity of α-glucosidase enzyme are: low free energy Gibs and low kinetic inhibition value. The Gibs free energy value (ΔG) obtained from the docking simulation showed that 13 cinnamic acid derivatives compounds had an affinity for the receptor α-glucosidase so it could act as α-glucosidase inhibitor. There were depicted significant interactions with the important residues of binding site.
Acknowledgments
We are very grateful for the scholarship that was given by The Ministry of Research and Technology for fiscal year of 2014-2018. In addition, I also am very thankful for all the support and help from Research Center for Chemistry – Indonesian Sciences and Institute.
Conflict of Interest
We declare no conflict of interest
References
- Frandsen, T. P. & Svensson, B. Plant Mol. Biol. 1988, 37, 1–13.
CrossRef - Nashiru, O., Koh, S., Lee, S. & Lee, D. J. Biochem. Mol. Biol. 2001, 34, 347–354.
- Bösenberg, L. H. & Zyl, D. G. van. J. Endocrinol. Metab. Diabetes South Africa.2008, 13, 3, 80-88.
CrossRef - Park, H. et al. Bioorg. Med. Chem. 2008, 16, 284–292.
CrossRef - Daviess, G. & Henrissat, B. Structure. 1995, 3, 853–859.
CrossRef - Laar, F. A. van De et al. Diabetes Care.2005, 28, 166–175.
- Luna, B. & Feinglos, M. N. Am. Fam. Phisician. 2011, 63, 1747–1756.
- Andrade-cetto, A. & Becerra-jim, J. J. Ethnopharmacol.2008, 116, 27–32.
CrossRef - Chiasson, J. et al. Lance. 2002, 359, 2072–2077.
CrossRef - Dabhi, A. S., Bhat, N. R. & Shah, M. J. J. Clin. Diagnostic Res.2013, 7, 3023–3027.
- Liu, Y. et al. Bioorg. Med. Chem. 2006, 14, 5683–5690.
CrossRef - Kalra, S. in J Pak Med Assoc. 2014, 64, 474–476.
- Phuwapraisirisan, P., Puksasook, T., Jong-aramruang, J. & Kokpol, U.. Bioorg. Med. Chem. Lett. 2008, 18, 4956–4958.
CrossRef - Adisakwattana, S. et al. Bioorg. Med. Chem. Lett. 2004, 14, 2893–2896.
CrossRef - Coleman, R. G., Carchia, M., Sterling, T., Irwin, J. J. & Shoichet, B. K. PLOS ONE. 2013, 8, 1–19.
CrossRef - Mohammed, K. et al. Eur. J. Med. Chem. 2014, 81, 245–252.
CrossRef - Yoshimizu, M., Tajima, Y., Matsuzawa, F. & Aikawa, S. Clin. Chim. Acta. 2008, 391, 68–73.
CrossRef - Sim, L. Disertation. 2010, 1–140, University of Toronto.
- Saqib, U. & Siddiqi, M. I. Int. J. Integr. Biol. 2009, 5, 13–19.
- Saqib, U. & Siddiqi, M. I. Int. J. Integr. Biol.2008, 2, 116–121.
- Azam, S. S., Uddin, R., Syed, A. A. S., Zaheer-ul-Haq. Med Chem Res.2012, 21, 1677–1683.
CrossRef - Walder, K. & McMillan, J. Inter. J. Obes Relat metab Disord. 2012, 26, 442–449.
- Mooradian, A. & Thurman, J. Drugs. 1999, 57, 19–29.
CrossRef - Hirschhorn, R., Huie, M. & Kasper, J. Proc.Natl.Acad.Sci . 2002, 99, 13642–13646.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


