Expeditious Synthesis and Spectroscopic Characterization of 2-Methyl-3-Substituted-Quinazolin-4(3H)-one Derivatives
1Department of Chemistry, Covenant University, CST, Canaanland, Km 10 Idiroko Road, P.M.B. 1023, Ota, Ogun State, Nigeria.
2Department of Chemistry, Georgia State University, Atlanta GA, 30302, USA.
Corresponding Author E-mail: ola.ajani@covenantuniversity.edu.ng
DOI : http://dx.doi.org/10.13005/ojc/330203
Download this article as:
![]()
Quinazoline and quinazolinone derivatives are well-known bioactive heterocycles owing to their therapeutic diversity and extensive medicinal application in drug design and pharmaceutics. A series of 2-methyl-3-substituted quinazolin-4(3H)-one derivatives 8a-q was herein synthesized from synthetic conversion of anthranilic acid to 2-methyl-4H-3,1-benzoxazi-4-one, 7 which was subsequently transformed to the targeted 2,3-disubstituted quinazolin-4(3H)-one derivatives 8a-q by reacting with some notable amino-containing moieties via an ameliorable pathway. The catalyst-free synthesis was successful achieved by careful reaction optimization study using solvent choice and reaction temperature variability as key parameters. The chemical structures of the synthesized compounds were confirmed by IR, UV, 1H-NMR, 13C-NMR and DEPT-135 as well as analytical data.
KEYWORDS:Solvent-dependent synthesis; spectral study; quinazolin-4(3H)-one; DEPT-135 analysis
Introduction
Heterocyclic compounds form the basis for pharmaceutical industry especially in the synthesis of drugs1. Also, most marketed drugs constitute of common structural units of heterocyclic compounds which could also be useful in the synthesis of biologically active agrochemicals 2,3. For instance, in the pharmaceutical industry, among the top two hundred branded drugs, we have more than 75% of them which have heterocyclic fragments in their structures4. One of these organic compounds of interest among the heterocycles is quinazoline otherwise known as 1,3-diazanaphthalene, benzo-1,3-diazine, benzopyrimidine which was first synthesized by Gabriel in 19035. Quinazoline can have different types of substituents attached to it, forming different derivatives of the parent quinazoline. Substituted quinazolines have properties which largely depend on: the nature and position of the substituent as well as type of conjugation in pyrimidine ring6. quinazolinones alkaloids form small but important group of naturally occurring bases which have been isolated from a number of different families e.g. seeds of P. nigellastium Bunge7. Quinazoline occurs mostly as alkaloid in root and leaves of some medicinal plants such as Dichroa febrifuga (also called Chinese quinine) which belongs to the Saxifragaceae family8. Numerous quinazoline derivatives have also be synthesized in order to achieve biologically active template which are essential in drug development. For instance, quinazoline motifs have been reported to exhibit diverse biological and pharmacological activities which include anticonvulsant9, anti-inflammatory10, antileishmanial11, antimalarial12, antioxidant13, antitubercular14, antiviral15 among others. There is vast amount of literature has been documented for the synthesis of quinazoline and quinazolinone on the catalyst supported synthesis of this crucial heterocyclic core, some of which are Ceryl ammonium nitrate (CAN) for synthesis of 116, Ru3(CO2)12 for 217, CuCl for 318, β-cyclodextrin/Montmorillonite for 419, iodine for 520 and Fe/HCl for 621 (Fig. 1). Some of these catalysts are costly to get. Hence, we have herein embarked on the synthesis of our targeted quinazolinone derivatives in the catalyst-free medium by engaging reaction optimization study using variation of solvents and temperature parameters.
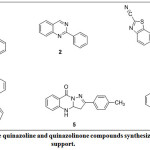 |
Figure 1: Some quinazoline and quinazolinone compounds synthesized by catalytic support. Click here to View Figure |
Experimental
All chemical compounds and reagents used were obtained from Sigma-Aldrich, USA except methanol and dichloromethane which were obtained from B.D.H. Chemicals, UK and were made available by Department of Chemistry, Covenant University. Solvents used were of analytical grade and were used directly without further purification. Melting points were determined in open capillary tubes using Stuart melting point apparatus and were uncorrected. The IR spectra were run in solid state using the Bruker FT-IR; while UV Spectrophotometric analyses of all the samples were run in ethanol, using UV-Genesys spectrophotometer in the Instrumentation Laboratory of Chemistry Department. The 1H-NMR and 13C-NMR of the compounds were run on Bruker NMR machine at 500 MHz AND 125 MHz respectively. Distortionless Enhancement Proton Transfer (DEPT-135) analysis was also investigated for effective characterization. The chemical shifts were measured with reference to tetramethylsilane (TMS) and the solvent used were CDCl3 and deuteriated DMSO depending on the solubility properties of the required compound. The elemental analysis (C, H, N) of the synthesized compounds were performed using a Flash EA 1112 CHN elemental analyzer. Results were found to be in good agreement with the calculated values (Table 3). All compounds were routinely checked by TLC on silica gel G plates and column chromatographic purifications were carried out on Merck silica gel F (Mesh 200-300). The progress of the reaction and the level of purity of the compounds were routinely checked by TLC on silica gel plates using different solvent system based on the variation of polarity of the product of interest and the developed plates were visualized under UV light. All drying was conducted at reduced pressure with DHG-9023A Vacuum Oven.
Synthesis of 2-Methyl-4H-3,1-Benzoxazin-4-one, 7
Acetic anhydride (30 ml) was added to anthranilic acid (30 g, 220 mmol), under continuous stirring for 5 mins at room temperature. An exothermic reaction was observed, then the reaction mixture was cooled in a refrigerator for 5 mins, after which it was heated under reflux and the progress of reaction was monitored by TLC using dichloromethane (DCM) as the eluting solvent. The reaction was completed in 6 h. The reaction mixture was then cooled to room temperature, filtered by suction and washed with few portion of cold water. It was then air-dried to afford 2-methyl-4H-3,1-benzoxazin-4-one, 7 as white crystalline solid in excellent yield. 1H-NMR (500 MHz, CDCl3) δH: 7.96-7.94 (d, J = 9.68 Hz, 1H of Ar-H), 7.77-7.73 (t, J = 9.68 Hz, 1H, Ar-H), 7.46-7.42 (t, J = 10.00 Hz, 1H, Ar-H), 7.42-7.40 (d, J = 10.00 Hz, 1H of Ar-H), 2.59 (s, 3H, CH3). 13C-NMR (125 MHz, DMSO-d6) δC: 168.77, (C=O), 140.37, 134.91, 130.74, 129.04, 127.82, 117.24, 114.52, 20.92 ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 129.11 (CH), 127.91 (CH), 117.14 (CH), 114.04 (CH), 20.92 (CH3) ppm. No Negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 214 (4.108), 250 (4.936), 263 (4.423). IR (KBr, cm–1) νmax: 3020 (C-H aromatic), 2929 (C-H aliphatic), 2846 (C-H aliphatic), 1743 (C=O of ester), 1620 (C=C), 1576 (C=N), 1388s (CH3 deformation), 1109 (C-O of alkoxyl), 975s (=C-H), 826 (C=C, out-of-plane bending), 757 (Ar-H, bending and ring puckering).
General Procedure for Synthesis of 2-Methyl-3-Substituted Quinazolin-4(3H)-ones, 8a-Q.
2-Methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) was dissolved in 10 ml of ethanol in a 250 ml round bottomed flask and the reaction mixture was allowed to stir for 5 mins at room temperature for complete dissolution, followed by the addition of the corresponding amino containing compounds (18.60 mmol). The mixture was then heated under reflux for the required number of periods as determined by the TLC monitoring. The reaction mixture was allowed to cool, filtered by suction and air-dried to afford moderate to excellent yields of various 2-methyl-3-substituted quinazolin-4(3H)-one derivatives, 8a-q.
Synthesis of 2,3-Dimethylquinazolin-4(3H)-one, 8a.
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with methyl amine (0.62 ml, 18.60 mmol) afforded 2,3-dimethylquinazolin-4(3H)-one, 8a. 1H-NMR (500 MHz, CDCl3) δH: 7.96-7.94 (d, J = 9.65 Hz, 1H of Ar-H), 7.77-7.73 (t, J = 9.00 Hz, 1H, Ar-H), 7.46-7.42 (t, J = 10.00 Hz, 1H, Ar-H), 7.42-7.40 (d, J = 9.30 Hz, 1H of Ar-H), 3.35 (s, 3H, CH3), 2.59 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, DMSO-d6) δC: 168.77, (C=O), 140.37, 134.91, 130.74, 129.04, 127.82, 117.24, 114.52, 25.26, 20.92 ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 129.11 (CH), 127.91 (CH), 117.14 (CH), 114.04 (CH), 25.24 (CH3), 20.92 (CH3) ppm. No Negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 212 (4.192), 251 (4.897), 293 (4.401). IR (KBr, cm–1) νmax: 3020 (C-H aromatic), 2929 (C-H aliphatic), 2845 (C-H aliphatic), 1691 (C=O of amide), 1620 (C=C), 1576 (C=N), 1445m, 1388s (CH3 deformation), 975s (=C-H), 826 (C=C, out-of-plane bending), 757 (Ar-H, bending and ring puckering).
Synthesis of 2-Methyl-3-Pentylquinazolin-4(3H)-one, 8b
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with pentylamine (1.74 ml, 18.60 mmol) afforded 2-methyl-3-pentylquinazolin-4(3H)-one, 8b. 1H-NMR (500 MHz, CDCl3) δH: 7.71-7.63 (dd, J1 = 10.30 Hz, J2 = 12.30 Hz, 2H, Ar-H), 7.28-7.26 (d, J = 12.3 Hz, 2H, Ar-H), 4.74-4.73 (m, 1H, CH), 3.72-3.69 (d, J = 10.20 Hz, 1H), 3.23-3.17 (t, J = 12.65 Hz, 1H), 2.41 (s, 3H, CH3-C=N), 2.18-2.15 (d, J = 12.65 Hz, 1H), 1.67-1.59 (m, 4H, 2 × CH2), 1.38-1.31 (t, J = 13.16 Hz, 3H, CH3-CH2). 13C-NMR (125 MHz, DMSO-d6) δC: 171.39 (C=O), 153.92, 145.83, 133.65, 127.42, 126.75, 126.25, 118.88, 40.98, 30.51, 27.38, 24.49 (CH3), 22.58, 14.51 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 133.65 (CH), 127.42 (CH), 126.75 (CH), 126.25 (CH), 24.49 (CH3), 14.51 (CH3). Negative signals are 40.98, 30.51, 27.38, 22.58 ppm. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 251 (4.915), 251 (4.979), 293 (4.440). IR (KBr, cm–1) νmax: 3038 (C-H aromatic), 2935 (C-H aliphatic), 2851 (C-H aliphatic), 1688 (C=O of amide), 1615 (C=C), 1576 (C=N), 1436m (CH2 deformation), 1388s (CH3 deformation), 977s (=C-H), 821(C=C, out-of-plane bending), 754 (Ar-H, bending and ring puckering), 658.
Synthesis of 3-Hexadecyl-2-Methylquinazolin-4(3H)-one, 8c
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with hexadecylamine (4.98 g, 18.60 mmol) afforded 3-hexadecyl-2-methylquinazolin-4(3H)-one, 8c. 1H-NMR (500 MHz, CDCl3) δH: 8.64-8.62 (d, J = 7.10 Hz, 1H of Ar-H), 7.95-7.94 (d, J = 7.60 Hz, 1H of Ar-H), 7.39-7.38 (t, J = 6.65 Hz, 1H of Ar-H), 6.99-6.96 (t, J = 6.95 Hz, 1H of Ar-H), 2.11 (s, 3H, CH3-C=N), 1.56-1.54 (t, J = 6.80 Hz, 2H, N-CH2-CH2), 1.22-1.11 (m, 28H, 2 × CH2), 0.86-0.83 (t, J = 6.80 Hz, 3H, CH3-CH2). 13C-NMR (125 MHz, DMSO-d6) δC: 174.38 (C=O), 168.33, 141.04, 132.45, 130.78, 121.80, 119.90, 39.69 (CH2), 31.65 (CH2), 29.52 (7 × CH2), 29.28 (CH2), 28.84 (CH2), 28.25 (CH2), 27.93 (CH2), 26.47 (CH2), 25.33 (CH3), 24.98 (CH2), 22.60 (CH2), 14.02 (CH3). DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 132.65 (CH), 130.98 (CH), 121.95 (CH), 119.96 (CH), 25.39 (CH3), 14.09 (CH3). Negative signals are 39.74 (CH2), 31.65 (CH2), 29.52 (7 × CH2), 29.28 (CH2), 28.84 (CH2), 28.25 (CH2), 27.93 (CH2), 26.47 (CH2), 24.98 (CH2), 22.60 (CH2). UV-Vis.: λmax in nm (log εmax mol-1cm-1): 209 (4.915), 248 (4.629), 299(3.833). IR (KBr, cm–1) νmax: 3433 (O-H), 3142 (C-H aromatic), 2918 (CH aliphatic), 2851 (CH aliphatic), 1690 (C=O of amide), 1660 (C=C), 1588 (C=N), 1437m (CH2 deformation), 1375s (CH3 deformation), 970s (=C-H), 825 (C=C, out-of-plane bending), 753 (Ar-H, bending and ring puckering).
Synthesis of (Z)-2-Methyl-3-(Octadec-9-En-1-Yl)Quinazolin-4(3H)-one, 8d
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with oleylamine (4.48 g, 18.60 mmol) afforded (Z)-2-methyl-3-(octadec-9-en-1-yl)quinazolin-4(3H)-one, 8d. 1H-NMR (500 MHz, CDCl3) δH: 8.13-8.12 (d, J = 5.05 Hz, 1H of Ar-H), 7.87-7.85 (t, J = 9.42 Hz, 1H, Ar-H), 7.46-7.42 (t, J = 10.00 Hz, 1H, Ar-H), 7.42-7.41 (d, J = 5.12 Hz, 1H of Ar-H), 5.42-5.40 (t, J = 12.80 Hz, 2H, CH2-CH=CH-CH2), 4.12 (t, J = 8.20 Hz, 2H, CH2-CH2), 2.58 (s, 3H, CH3-C=N), 2.18-2.15 (m, 4H, 2 × CH2), 1.67 (m, 2H, CH2), 1.31-1.29 (2H, CH2-CH2-CH3), 1.29-1.25 (m, 20H, 10 × CH2), 0.95 (t, J = 8.54 Hz, 3H, CH3-CH2). 13C-NMR (125 MHz, DMSO-d6) δC: 171.21 (C=O), 154.61, 146.92, 133.54, 130.73 (2 × CHolefin), 127.42, 126.84, 126.62, 120.92, 40.53 (CH2), 31.80 (CH2), 30.91 (CH2), 29.90 (2 × CH2), 29.71 (4 × CH2), 29.33 (2 × CH2), 27.84 (2 × CH2), 26.70 (CH2), 22.90 (CH3), 22.61 (CH2), 14.33 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 133.54 (CH), 130.73 (2 × CHolefin), 127.42 (CH), 126.84 (CH), 126.62 (CH), 22.90 (CH3), 14.33 (CH3) ppm. Negative signals are 40.53 (CH2), 31.80 (CH2), 30.91 (CH2), 29.90 (2 × CH2), 29.71 (4 × CH2), 29.33 (2 × CH2), 27.84 (2 × CH2), 26.70 (CH2), 22.61 (CH2) ppm. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 215 (5.434), 251(5.321), 293(4.762). IR (KBr, cm–1) νmax: 3038 (C-H aromatic), 3010 (C-H alkene), 2935 (C-H aliphatic), 2851 (C-H aliphatic), 1688 (C=O of amide), 1615 (C=C), 1576 (C=N), 1436m (CH2 deformation), 1388s (CH3 deformation), 977s (=C-H), 821 (C=C, out-of-plane bending), 754 (Ar-H, bending and ring puckering), 658.
Synthesis of 3-Cyclohexyl-2-Methylquinazolin-4(3H)-one, 8e
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with cyclohexylamine (2.13 ml, 18.60 mmol) afforded 3-cyclohexyl-2-methylquinazolin-4(3H)-one, 8e. 1H-NMR (500 MHz, CDCl3) δH: 8.03-8.01 (d, J = 5.09 Hz, 1H of Ar-H), 7.86-7.82 (t, J = 9.45 Hz, 1H, Ar-H), 7.45-7.42 (t, J = 10.00 Hz, 1H, Ar-H), 7.38-7.41 (d, J = 5.12 Hz, 1H of Ar-H), 3.54-3.51 (quintet, J = 4.50 Hz, 1H, CH2-CH-CH2), 2.59 (s, 3H, CH3-C=N), 1.77-1.63 (m, 4H, 2 × CH2), 1.50-1.47 (quintet, J = 7.54 Hz, 2H, CH2-CH2-CH2), 1.21-1.13 (m, 4H, 2 × CH2). 13C-NMR (125 MHz, CDCl3) δC: 170.21 (C=O), 154.11, 146.12, 133.25, 127.80, 126.91, 126.36, 120.54, 63.12, 31.42 (2 × CH2), 25.80, 24.63 (2 × CH2), 22.98 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 133.25, 127.80, 126.91, 126.36 (CHs), 22.98 (CH3) ppm. Negative signals are 31.42 (2 × CH2), 25.80, 24.63 (2 × CH2) ppm. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 215 (5.273), 251 (5.059), 296 (4.545). IR (KBr, cm–1) νmax: 3040 (C-H aromatic), 2919 (CH aliphatic), 2845 (CH aliphatic), 1690 (C=O of amide), 1620 (C=C), 1585 (C=N), 1435m (CH2 deformation), 1370s (CH3 deformation), 970s (=C-H), 830 (C=C, out-of-plane bending), 755 (Ar-H, bending and ring puckering), 658.
Synthesis of 2-Methyl-3-Phenylquinazolin-4(3H)-one, 8f
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with aniline (1.70 ml, 18.60 mmol) afforded 2-methyl-3-phenylquinazolin-4(3H)-one, 8f. 1H-NMR (500 MHz, CDCl3) δH: 7.61-7.59 (d, J = 10.00 Hz, 2H of Ar-H), 7.24-7.21 (m, 5H, Ar-H), 7.10-7.08 (m, 2H, Ar-H), 2.40 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, DMSO-d6) δC: 168.77 (C=O), 139.75, 137.18, 132.91, 132.07, 129.33, 129.15, 127.94, 125.64, 125.00, 123.40, 122.17, 121.18, 118.64, 22.39 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 129.33 (CH), 129.11 (CH), 125.50 (CH), 125.00 (CH), 123.40 (CH), 122.17 (CH), 121.18 (CH), 118.64 (CH), 22.35 (CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 218 (5.188), 302(4.403). IR (KBr, cm–1) νmax: 3108 (C-H aromatic), 3041 (C-H aromatic), 2929 (CH aliphatic from CH3), 1690 (C=O of amide), 1610 (C=C), 1575 (C=N), 1450, 1375 (CH3 deformation), 970s (=C-H), 831 (C=C, out-of-plane bending), 755 (Ar-H, bending and ring puckering), 651.
Synthesis of 3-(4-Chlorophenyl)-2-Methylquinazolin-4(3H)-one, 8g
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with 4-chloroaniline (2.38 g, 18.60 mmol) afforded 3-(4-chlorophenyl)-2-methylquinazolin-4(3H)-one, 8g. 1H-NMR (500 MHz, CDCl3) δH: 8.68-8.67 (d, J = 5.40 Hz, 1H of Ar-H), 8.09-8.08 (d, J = 10.09. Hz, 1H, Ar-H), 7.77-7.60 (m, 2H, ArH), 7.54-7.52 (d, J = 10.00 Hz, 2H, Ar-H), 7.32-7.29 (d, J = 10.50 Hz, 1H, Ar-H), 7.09-7.08 (d, J = 5.00 Hz, 1H, Ar-H), 2.23 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, DMSO-d6) δC: 168.77, (C=O), 140.37, 134.91, 130.74, 129.04, 127.82, 117.24, 114.52, 25.26, 20.92 ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 129.11 (CH), 127.91 (CH), 117.14 (CH), 114.04 (CH), 25.24 (CH3), 20.92 (CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 218 (5.369), 245 (5.287), 299 (4.690). IR (KBr, cm–1) νmax: 3477, 3378 (N-H, two bands), 3191 (C-H aromatic), 3113 (C-H aromatic), 2925 (CH aliphatic from CH3), 1693 (C=O of amide), 1650, 1606 (C=C), 1582 (C=N), 1452, 1377 (CH3 deformation), 968s (=C-H), 833 (C=C, out-of-plane bending), 805 (C-Cl stretching), 756 (Ar-H, bending and ring puckering), 655.
Synthesis of 3-(4-Bromophenyl)-2-Methylquinazolin-4(3H)-one, 8h
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with 4-bromoaniline (3.20 g, 18.60 mmol) afforded 3-(4-bromophenyl)-2-methylquinazolin-4(3H)-one, 8h. 1H-NMR (500 MHz, DMSO-d6) δH: 1H-NMR (500 MHz, CDCl3) δH: 8.18-8.14 (d, J = 5.80 Hz, 1H of Ar-H), 8.02-7.98 (d, J = 7.86 Hz, 1H, Ar-H), 7.74-7.58 (m, 2H, ArH), 7.53-7.51 (d, J = 10.00 Hz, 2H, Ar-H), 7.28-7.25 (d, J = 10.54 Hz, 1H, Ar-H), 7.03-7.01 (d, J = 5.00 Hz, 1H, Ar-H), 2.25 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, DMSO-d6) δC: 168.75 (C=O), 140.22, 137.40, 134.82, 131.15, 131.01, 130.32, 129.00, 128.13, 127.71, 125.31, 122.14, 117.04, 114.11, 25.12 ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 131.15, 131.01, 130.32, 129.00, 128.13, 127.71, 117.04, 114.11 (CHs), 25.12 (CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 245 (5.405), 206 (5.392), 296 (4.719). IR (KBr, cm–1) νmax: 3188 (C-H aromatic), 3110 (C-H aromatic), 2925 (CH aliphatic from CH3), 1688 (C=O of amide), 1600 (C=C), 1582 (C=N), 1449, 1373 (CH3 deformation), 968s (=C-H), 835 (C=C, out-of-plane bending), 755 (Ar-H, bending and ring puckering), 650, 505 (C-Br stretching).
Synthesis of 3-(2-Aminophenyl)-2-Methylquinazolin-4(3H)-one, 8i
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with o-phenylenediamine (2.02 g, 18.60 mmol) afforded 3-(2-aminophenyl)-2-methylquinazolin-4(3H)-one, 8i. 1H-NMR (500 MHz, DMSO-d6) δH: 1H-NMR (500 MHz, CDCl3) δH: 8.10-8.07 (d, J = 5.70 Hz, 1H of Ar-H), 7.88-7.85 (d, J = 7.80 Hz, 1H, Ar-H), 7.72-7.66 (m, 2H, ArH), 7.44-7.40 (d, J = 10.05 Hz, 1H, Ar-H), 7.28-7.25 (d, J = 10.54 Hz, 1H, Ar-H), 7.11-7.07 (m, 1H, Ar-H), 6.42 (s, 2H, NH2), 2.35 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, DMSO-d6) δC: 171.05 (C=O), 154.60, 140.22, 137.33, 134.81, 131.00, 130.32, 129.00, 128.13, 127.71, 125.31, 122.14, 117.04, 114.11, 25.20 ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 131.00, 130.32, 129.00, 128.13, 127.71, 125.31, 117,04, 114.11 (CHs), 25.20 (CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 206 (4.997), 248 (4.598), 290 (4.223). IR (KBr, cm–1) νmax: 3411 (N-H), 3255 (N-H), 3015 (C-H aromatic), 2929 (C-H aliphatic), 2854 (C-H aliphatic), 1949, 1685 (C=O of amide), 1615 (C=C aromatic), 1575 (C=N), 1450, 1385 (CH3 deformation), 1245 (C-N), 968s (=C-H), 869 (C=C, out-of-plane bending), 760 (Ar-H, bending and ring puckering), 650.
Synthesis of 3,3-(1,2-Phenylene)Bis(2-Methylquinazolin-4(3H)-one, 8j
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with o-phenylene diamine (1.01 g, 9.30 mmol, 0.5 equiv.) afforded 3,3-(1,2-phenylene)bis(2-methyl quinazolin-4(3H)-one, 8j. 13C-NMR (125 MHz, CDCl3) δC: 171.05 (2 × C=O), 144.12 (2 × C), 140.91 (2 × C), 134.21 (2 × C), 133.88 (2 × C), 127.31 (2 × C), 126.71 (2 × C), 126.60 (2 × C), 123.18 (2 × C), 120.77 (2 × C), 114.33 (2 × C), 25.12 (2 × CH3). DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 127.31 (2 × CH), 126.71 (2 × CH), 126.60 (2 × CH), 123.18 (2 × CH), 120.77 (2 × CH), 114.33 (2 × CH), 25.12 (2 × CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 206 (4.909), 251(4.568), 296(4.130).IR (KBr, cm–1) νmax: 3050 (C-H aromatic), 3011 (C-H aromatic), 2925 (C-H aliphatic), 2857 (C-H aliphatic), 1688 (C=O of amide), 1620 (C=C aromatic), 1576 (C=N), 1451, 1386 (CH3 deformation), 1247 (C-N), 969s (=C-H), 879 (C=C, out-of-plane bending), 760 (Ar-H, bending and ring puckering), 655.
Synthesis of 3-(2-Mercaptophenyl)-2-Methylquinazolin-4(3H)-one, 8k
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with 2-aminothiophenol (2.00 ml, 18.60 mmol) afforded 3-(2-mercaptophenyl)-2-methylquinazolin-4(3H)-one, 8k. 1H-NMR (500 MHz, CDCl3) δH: 8.11-8.07 (d, J = 5.68 Hz, 1H of Ar-H), 7.89-7.87 (d, J = 7.80 Hz, 1H, Ar-H), 7.70-7.65 (m, 2H, ArH), 7.46-7.43 (d, J = 10.00 Hz, 1H, Ar-H), 7.7-7.22 (d, J = 10.50 Hz, 1H, Ar-H), 7.10-7.05 (m, 1H, Ar-H), 3.42 (s, 1H, SH), 2.33 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, CDCl3) δC: 171.11 (C=O), 144.38, 141.09, 137.13, 134.27, 131.09, 130.88, 129.20, 128.11, 127.23, 125.73, 122.10, 117.04, 114.11, 25.23 ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 131.09, 130.88, 129.20, 128.11, 127.23, 125.73, 118.23, 114.11 (CHs), 25.23 (CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 218 (5.310), 248 (4.969), 296 (4.497). IR (KBr, cm–1) νmax: 3112 (C-H aromatic), 3029 (C-H aromatic), 2925 (CH aliphatic from CH3), 2556w (S-H of thiol, sharp and weak), 1690 (C=O of amide), 1610 (C=C), 1585 (C=N), 1452, 1375 (CH3 deformation), 970s (=C-H), 833 (C=C, out-of-plane bending), 756 (Ar-H, bending and ring puckering), 651.
Synthesis of 3,3′-(1,4-Phenylene)Bis(2-Methylquinazolin-4(3H)-one), 8l
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with o-phenylenediamine (1.01 g, 9.30 mmol, 0.5 equiv.) afforded 3,3′-(1,4-phenylene)bis(2-methyl quinazolin-4(3H)-one), 8l. 1H-NMR (500 MHz, CDCl3) δH: 8.10-8.07 (d, J = 5.60 Hz, 2H of 2 × Ar-H), 7.89-7.87 (d, J = 7.82 Hz, 2H, 2 × Ar-H), 7.70-7.65 (m, 4H, 2 × Ar-H), 7.36-7.34 (d, J = 10.00 Hz, 4H, 2 × Ar-H), 2.26 (s, 6H, 2 × CH3). 13C-NMR (125 MHz, CDCl3) δC: 170.23 (2 × C=O), 145.21 (2 × C), 137.19 (2 × C), 134.02 (2 × C), 130.16 (2 × C), 129.38 (2 × C), 122.77 (2 × C), 117.09 (2 × C), 25.22 (2 × CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 137.19 (2 × CH), 134.02 (2 × CH), 129.38 (2 × CH), 122.77 (2 × CH), 25.22 (2 × CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 218(4.793), 251(4.545), 305(3.851). IR (KBr, cm–1) νmax: 3056 (C-H aromatic), 3022 (C-H aromatic), 2925 (CH aliphatic from CH3), 1688 (C=O of amide), 1600 (C=C), 1575 (C=N), 1450, 1370 (CH3 deformation), 969s (=C-H), 831 (C=C, out-of-plane bending), 755 (Ar-H, bending and ring puckering), 650.
Synthesis of 1-(2-Methyl-4-oxoquinazolin-3(4H)-Yl)urea, 8m
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with semicarbazone (1.39 g, 18.60 mmol) afforded 1-(2-methyl-4-oxoquinazolin-3(4H)-yl)urea, 8m.
1H-NMR (500 MHz, CDCl3) δH: 10.96 (s, 1H, NH), 8.08-8.06 (d, J = 9.56 Hz, 2H, Ar-H), 7.81-7.68 (m, 2H, Ar-H), 7.08 (s, 2H, NH2), 2.22 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, CDCl3) δC: 173.21 (C=O), 171.81 (C=O), 151.18, 135.00, 132.27, 127.43, 117.04, 116.82, 109.99, 25.23 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 135.00 (CH), 132.27 (CH), 117.04 (CH), 116.82 (CH), 25.23 (CH3) ppm. No Negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 221 (5.349), 302 (4.464), 545 (3.079). IR (KBr, cm–1) νmax: 3415 (N-H), 3260 (N-H), 3176 (C-H aromatic), 2921 (C-H aliphatic), 2850 (C-H aliphatic), 1949, 1688 (C=O of amide), 1606 (C=C aromatic), 1584 (C=N), 1453, 1383 (CH3 deformation), 1248 (C-N), 967s (=C-H), 879 (C=C, out-of-plane bending), 766 (Ar-H, bending and ring puckering).
Synthesis of 3-Amino-2-Methylquinazolin-4(3H)-one, 8n
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with hydrazine hydrate (0.91 ml, 18.60 mmol) afforded 3-amino-2-methylquinazolin-4(3H)-one, 8n. 1H-NMR (500 MHz, CDCl3) δH: 7.92-7.90 (d, J = 9.56 Hz, 2H, Ar-H), 7.30-7.27 (m, 2H, Ar-H), 6.50 (s, 2H, NH2), 2.07 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, CDCl3) δC: 173.21 (C=O), 151.18, 135.00, 132.27, 122.89, 117.04, 116.82, 109.99, 20.78 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: +ve signals are 135.00 (CH), 132.27 (CH), 117.04 (CH), 116.82 (CH), 20.78 (CH3) ppm. No –ve signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 230 (5.443), 245(5.439), 320(5.089). IR (KBr, cm–1) νmax: 3479 (N-H of NH2), 3394 (N-H), 3185 (C-H aromatic), 3049 (C-H aromatic), 2921 (C-H aliphatic), 1948 (C=C asym. stretching), 1689 (C=O), 1652 (C=C), 1581 (C=N), 1453, 1376 (CH3 deformation), 1245 (C-N), 968s (=C-H), 880 (C=C, out-of-plane bending), 756 (Ar-H, bending and ring puckering), 660.
Synthesis of 3-(2,4-Dinitrophenylamino)-2-Methylquinazolin-4(3H)-one, 8o
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with 2,4-dinitrophenylhydrazine (2,4-DNP) (3.59 g, 18.30 mmol) afforded 3-(2,4-dinitrophenylamino)-2-methylquinazolin-4(3H)-one, 8o. 1H-NMR (500 MHz, CDCl3) δH: 9.01 (s, 1H, Ar-H), 8.52-8.50 (d, J = 8.00 Hz, 2H, Ar-H), 8.04-8.01 (d, J = 9.22 Hz, 1H, Ar-H), 7.45-7.40 (d, J = 9.22 Hz, 1H, Ar-H), 7.38-7.36 (d, J = 8.00 Hz, 2H, Ar-H), 7.11.7.15 (m, 2H, Ar-H), 5.60 (s, 1H, NH), 2.55 (s, 3H, CH3). 13C-NMR (125 MHz, CDCl3) δC: 171.21 (C=O), 151.18, 146.33, 140.12, 135.15, 138.19, 132.03, 129.70, 126.73, 122.89, 120.13, 117.84, 116.29, 109.99, 24.78 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 146.33, 140.12, 138.19, 135.15, 132.03, 117.84, 116.29 (CHs), 24.78 (CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 218 (4.650), 248(4.354), 347(4.130). IR (KBr, cm–1) νmax: 3420 (N-H), 3365 (N-H), 3100 (C-H aromatic), 3045 (C-H aromatic), 2925 (C-H aliphatic), 2850 (C-H aliphatic), 1945 (C=C asym. stretching), 1690 (C=O), 1622 (C=C), 1585 (C=N), 1535 (N-O Asymmetric stretching of NO2), 1455, 1376 (CH3 deformation), 1315 (N-O symmetric stretching of NO2),1245 (C-N), 970s (=C-H), 881 (C=C, out-of-plane bending), 755 (Ar-H, bending and ring puckering), 665.
Synthesis of 2-Methyl-3-(Naphthalen-2-Yl)Quinazolin-4(3H)-one, 8p
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with α-naphthylamine (2.67 g, 18.60 mmol) afforded 2-methyl-3-(naphthalen-2-yl)quinazolin-4(3H)-one, 8p. 1H-NMR (500 MHz, CDCl3) δH: 8.07-8.05 (d, J = 5.88 Hz, 1H, Ar-H), 8.03-8.01 (d, J = 5.68 Hz, 1H, Ar-H), 7.69-7.67 (d, J = 8.00 Hz, 1H, Ar-H), 7.55-7.52 (m, 3H, Ar-H), 7.47-7.43 (m, 2H, Ar-H), 7.28-7.26 (d, J = 10.29 Hz, 1H, Ar-H), 2.41 (s, 3H, CH3-C=N). 13C-NMR (125 MHz, DMSO-d6) δC: 168.74 (C=O), 144.21, 139.70, 137.43, 132.61, 132.01, 129.36, 129.14, 127.89, 126.82, 126.12, 125.57, 125.02, 124.71, 123.40, 122.17, 121.18, 118.64, 22.40 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 139.70, 132.61, 129.36 (CH), 129.14 (CH), 126.81, 126.12, 125.57 (CH), 125.02 (CH), 123.40 (CH), 121.18 (CH), 118.64 (CH), 22.40 (CH3) ppm. No negative signals. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 224 (5.349), 242 (5.444), 305 (4.971).IR (KBr, cm–1) νmax: 2919 (C-H aliphatic), 2084, 1690 (C=O), 1610 (C=C aromatic), 1485, 1369 (CH3 deformation), 1246 (C-N), 960s (=C-H), 829 (C=C, out-of-plane bending), 758 (Ar-H, bending and ring puckering), 654.
Synthesis of 2-Methyl-3-(3-(Trimethoxysilyl)Propyl)Quinazolin-4(3H)-one, 8q
Treatment of 2-methyl-4H-3,1-benzoxazin-4-one, 7 (3.00 g, 18.60 mmol) with 3-amino propyltrimethoxysilane (3.25 ml, 18.60 mmol) afforded 2-methyl-3-(3-(trimethoxysilyl) propyl)quinazolin-4(3H)-one, 8q. 1H-NMR (500 MHz, CDCl3) δH: 7.94-7.92 (d, J = 9.56 Hz, 1H, Ar-H), 7.37-7.35 (d, J = 8.00 Hz, 1H, Ar-H), 7.15-7.09 (m, 2H, Ar-H), 4.14-4.11 (t, J = 5.44 Hz, 2H, N-CH2-CH2), 3.52 (s, 9H, 3 × CH3, Si-O-CH3), 2.52 (s, 3H, CH3-C=N), 1.68-1.65 (m, 2H, CH2), 1.22-1.25 (t, J = 7.56 Hz, 2H, Si-CH2-CH2). 13C-NMR (125 MHz, CDCl3) δC: 171.31 (C=O), 135.20, 132.27, 127.45, 126.25, 122.89, 117.04, 51.32 (3 × O-CH3), 46.27 (CH2), 23.48 (CH2), 16.61 (S-CH2), 22.49 (CH3) ppm. DEPT 135 (125 MHz, CDCl3) δC: Positive signals are 132.27, 127.45, 126.25, 117.04 (CHs), 116.82 (CH), 51.32 (3 × CH3), 22.49 (CH3) ppm. Negative signals are 46.27 (CH2), 23.48 (CH2), 16.61 (CH2) ppm. UV-Vis.: λmax in nm (log εmax mol-1cm-1): 215 (4.924), 248 (4.691), 299(4.196). IR (KBr, cm–1) νmax: 3101 (C-H aromatic), 3022 (C-H aromatic), 2929 (C-H aliphatic), 2855 (C-H aliphatic), 1945 (C=C asym. stretching), 1690 (C=O), 1620 (C=C), 1575 (C=N), 1450, 1377 (CH3 deformation), 1245 (C-N), 1050 (Si-OCH3, str. & brd.), 973s (=C-H), 885 (C=C, out-of-plane bending), 755 (Ar-H, bending and ring puckering), 668.
Result and Discussion
Chemistry
Quinazolinone derivatives are important structural scaffolds found in various library of biologically active compounds which are medicinally useful agents in formulation, discovery of drugs and medicinal chemistry research. Hence, in the continuation of our effort on the synthesis of N-heterocyclic motifs1,2,4, we have herein achieved expeditious synthesis of 2-methyl-3-substituted quinazoline-4(3H)-one derivatives 8a-q, in a two steps technique. The first step of the synthesis in this present research work, involved the reaction of anthranilic acid with acetic anhydride according to a known procedure reported by Hassan et al.22 in modified version to afford reactive intermediate 2-methyl-4H-3,1-benzoxazin-4-one, 7 which was used as the reactive intermediate for the second step. The completion of the reaction was monitored with thin layer chromatography (TLC) using dichloromethane: methanol (9.5:0.5) as the eluting solvent system. The solid formed was filtered via suction and air-dried in order to afford 2-methyl-4H-3,1-benzoxazin-4-one 7 in good yield (Scheme 1). However, before this intermediate was taking further to achieve all targeted compounds 8a-q, the reaction optimization study was carried out via the synthesis of 8a which was the first member of this series. Preliminary optimization of reaction conditions was done by considering the effect of solvent type and reaction temperature for the effective synthesis of 8a being the first and simplest 2-methyl-3-substitutedquinazolin-4(3H)-one since the substituent at position-3 of its template was a methyl group. The synthesis of 8a was attempted in four different entries via thermal reaction of 2-methyl-4H-3,1-benzoxazine-4-one with methyl amine at reflux condition for 6 h using three solvents which were butanol, ethanol and dichloromethane as well as under solvent-free media (Table 1). This was done for comparative study of the efficiency of each solvent system for obtaining 8a. It was observed that the reaction in ethanol gave the most suitable condition for the synthesis of 8a, since it resulted in the highest yield (79.78%), followed by butanol (55.83%) and then dichloromethane which gave the lowest yield (45.35%). However, it was interesting to note that solvent-less medium resulted in no isolable product but reverted back to the staring material as indicated by the observation found in the TLC spotting which showed the presence of the starting material without any new spot for the product, while the solvent free medium gave the least yield (18.21%) as most of the starting material was not effectively converted to product as noticed in the TLC spotting monitoring. and therefore it was used in all subsequent experiments. The effect of temperature was studied by carrying out the synthesis of 8a at different temperatures; 40, 70, 100 and 130 oC (Table 2) using ethanol solvent which was unveiled as the best solvent during the solvent-dependent optimization study and the result was as shown in Table 2. At refluxing reaction temperature of 40 oC, the yield of 8a was 18.53% obtained; and as the temperature increased to 70 oC, the percentage yield of 8a also increased to 55.82%, while the highest yield of 8a was observed to be 79.78% at a reacting temperature of 100 oC. However, the percentage yield decreased sharply to 35.48% when the reaction was experimented at temperature of 130 oC (Table 2). Hence, it was concluded that the optimum temperature for the expeditious synthesis of 8a was 100 oC and in the presence of ethanol solvent. Therefore, the same reaction condition was subsequently use for the synthesis of the remaining sixteen 2-methyl-3-substituted quinazolin-4(3H)-one motifs, 8b-q through synthetic modification of the -COOC end by condensing it with various cheap and readily available amino containing compounds in the presence of ethanol at 100 oC (Scheme 2). The completion of the reaction was confirmed after 6 h of refluxing using TLC spotting, then the solution was left to stand overnight in freezer chest for proper crystallization to afford the targeted 2-methyl-3-substituted quinazolin-4(3H)-one derivative (8a-q) in appreciable yields.
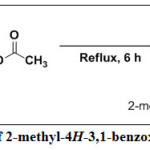 |
Scheme 1: Synthesis of 2-methyl-4H-3,1-benzoxazin-4-one, 7. Click here to View Scheme |
Table 1: Investigation of solvent-dependent synthesis of (8a) under reflux condition
| Entry | Solvent |
Time / h |
Yield / % |
| 1 | Ethanol |
6 |
79.78 |
| 2 | Butanol |
6 |
55.83 |
| 3 | Dichloromethane |
6 |
45.35 |
| 4 | Solvent free |
6 |
NIPO |
NIPO = No Isolable Product Observed
Table 2: Investigation of temperature-dependent synthesis of (8a) in ethanol
|
Entry |
Solvent |
Temperature / oC |
Yield / % |
| 1 | Ethanol |
130 |
35.48 |
| 2 | Ethanol |
100 |
79.78 |
| 3 | Ethanol |
70 |
55.62 |
| 4 | Ethanol |
40 |
18.53 |
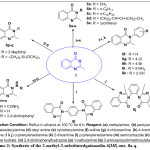 |
Scheme 2: Synthesis of the 2-methyl-3 substitut edquinazolin-4(3H)-one, 8a-q. Click here to View Scheme |
Physico-chemical Analysis
The result of the physico-chemical parameters of the synthesized compounds 8a-q was as shown in Table 3. These parameters investigated include molecular weight, Rf value, melting point, percentage yield, sample colour and elemental analytical result for carbon, hydrogen and nitrogen. The molecular weights of the targeted compounds ranged 174.08 for compound 8a to 410.64 for compound 8d. The Rf value for the targeted compounds varied from 0.36 for compound 8l to 0.91 for compound 8n. The Rf values did not only showed the level of purity for each compound but also established the polarity of these targeted compounds. The melting point of the targeted compounds 8a-q was also presented in Table 3. It showed that compound 8b and 8m had the highest melting point value of > 300°C while compound 8f has the lowest melting point value of 117-121°C. The melting points of other compounds, which include 8i, 8j, 8k and 8l, were not determined due to the fact that they were oily and liquid at room temperature. The inability of compound 8m to melt at the highest permissible limit (300°C) of the melting point apparatus might be due to the presence of the additional side chain amide which had tendency to exhibit hydrogen bong formation. The next parameter is the percentage yield which varied from 49.90% for compound 8i to 99.54% for compound 8n. The colour of the synthesized compounds was determined via virtual observation to be seven different colours which are brown, cream, black, yellow, purple, red and white. Compounds 8a, 8b, 8d, 8e, 8g, 8h, 8n and 8q were observed to be brown in colour, followed by compound 8c with cream colour while compounds 8f was black in colour. Compounds 8i, 8j and 8k were observed to be yellow in colour followed by compounds 8l, 8q and 8q which were purple whereas, compound 8m was noticed to be white in colour. The result of the elemental analytical data for the carbon, hydrogen and nitrogen of the intermediate 7 and the targeted compounds 8a-q was the last parameter presented in Table 3. The result of elemental analysis did not only correlate well with the molecular masses of all synthesized compounds but also showed a consistent minimum difference of not more than ±0.39 between % calculated and % found for the carbon, hydrogen and nitrogen of the targeted products 8a-q.
Table 3: Result of the physico-chemical parameters of synthesized compounds, 8a-q.
|
Comp. No |
Mol. Form. (Mol. Wt.) | RfValue | MeltingPt. (oC) | Yield (%) | Sample Colour | Elemental Analysis %Calcd. (% Found) | ||
| C | H |
N |
||||||
| 8a | C10H10N2O (174.08) | 0.62a | 285 | 79.78 | Brown | 68.95 (69.01) | 5.79 (5.73) | 9.18 (9.22) |
| 8b | C14H18N2O (230.31) | 0.83a | > 300 | 83.75 | Brown | 73.01 (72.97) | 7.88 (8.06) | 12.16 (12.11) |
| 8c | C25H40N2O (384.60) | 0.62a | 278-281 | 98.10 | Cream | 78.07 (77.97) | 10.48 (10.31) | 7.28 (7.14) |
| 8d | C27H42N2O (410.64) | 0.58a | 267-272 | 85.65 | Brown | 78.97 (79.04) | 10.31 (10.28) | 6.82 (7.00) |
| 8e | C15H18N2O (242.32) | 0.89a | 178-181 | 57.43 | Brown | 74.35 (74. 28) | 7.49 (7.54) | 11.56 (11.61) |
| 8f | C15H12N2O (236.27) | 0.71a | 117-121 | 61.57 | Black | 76.25 (76.33) | 5.12 (4.97) | 11.86 (12.01) |
| 8g | C15H11N2OCl (270.71) | 0.72b | 292-296 | 94.78 | Brown | 66.55 (66.41) | 4.10 (3.97) | 10.35 (10.42) |
| 8h | C15H11N2OBr (315.16) | 0.82 b | 285-287 | 95.13 | Brown | 57.16 (57.22) | 3.52 (3.66) | 8.89 (9.02) |
| 8i | C15H13N3O (251.28) | 0.68a | ND | 49.90 | Yellow | 71.70 (71.55) | 5.21 (5.08) | 16.72 (16.61) |
| 8j | C24H18N4O2 (394.43) | 0.66a | ND | 51.37 | Yellow | 73.08 (72.97) | 4.60 (4.47) | 14.20 (14.32) |
| 8k | C15H12N2OS (268.33) | 0.60a | ND | 78.56 | Yellow | 67.14 (66.98) | 4.51 (4.39) | 10.44 (10.54) |
| 8l | C24H18N4O2 (394.43) | 0.36a | ND | 60.47 | Purple | 73.08 (72.94) | 4.60 (4.51) | 14.20 (14.37) |
| 8m | C10H10N4O2 (218.21) | 0.55a | > 300 | 71.97 | White | 55.04 (54.97) | 4.62 (4.55) | 25.68 (25.75) |
| 8n | C9H9N3O (175.19) | 0.91a | 138-140 | 99.54 | Brown | 61.70 (61.88) | 5.18 (4.99) | 23.99 (24.08) |
| 8o | C15H11N5O5 (341.28) | 0.56 b | 153-155 | 74.84 | Red | 52.79 (52.61) | 3.25 (3.31) | 20.52 (20.67) |
| 8p | C19H14N2O (286.33) | 0.72a | 213-215 | 96.44 | Purple | 79.70 (79.62) | 4.93 (5.02) | 9.78 (9.87) |
| 8q | C19H14N2O (322.43) | 0.58a | 258-261 | 81.52 | Brown | 55.88 (56.02) | 6.88 (7.04) | 8.69 (8.75) |
Comp. No = Compound No; Mol. Form. = Molecular Formular; Mol. Wt. = Molecular Weight; ND = Not Determined. Solvent System: aDCM : MeOH (9.5:0.5); bDCM : MeOH (9:1).
Spectroscopic Characterization
The results of the electronic transition using uv spectroscopy was investigated in ethanol solution and the results is a presented in the experimental section showing the wavelength (λmax) and the log of molar absorptivity (Log εmax). The infrared analysis was carried out using Bruker Fourier-Transform Infrared (FT-IR) and the various absorption bands from stretching and bending vibrational frequencies were duly reported. The results of the infrared spectral data of all the synthesized compounds were as presented in the experimental section. The 1H-NMR and 13C-NMR as well as DEPT-135 (Distortionless Enhancement by Polarization Transfer) of the reactive intermediate and targeted quinazolin-4-one motifs 8a-q were run in either DMSO-d6 or CDCl3 depending on the solubility behaviour of each of the compound to be analyzed per time. The 1H-NMR spectrum of precursor 7 was run in CDCl3. It showed the presence of four aromatic protons between δ 7.96 and 7.40 ppm. They are 1H doublet at δ 7.77-7.73 ppm which was in the neighborhood of 1H triplet since they both possessed the same coupling constant of 9.68 Hz. Similarly, the remaining two aromatic protons were observed as 1H doublet at δ 7.46-7.42 ppm and 1H triplet at δ 7.42-7.40 ppm with coupling constant of 10.00 Hz. The 3H of methyl group was observed up-field of TMS as a singlet at δ 2.59 ppm. Apart from all other signals in 1H NMR of compound 7 which were replicated in representative targeted product 8a, it is crucial to note that the 1H NMR spectrum of 8a also showed one addition signal at chemical shift value of δ 3.35 ppm. This authenticated the insertion of methyl group at the position 3 of compound 7 to afford 8a. It was worthy to note that these values fall within the range for aromatic protons (8.28-7.09 ppm) earlier reported by Ajani et al., (2010) who investigated the microwave assisted synthesis of a series of novel 2-quinoxalinone-3-hydrazone23 which also belong to benzodiazine family like quinazolinone but with its own nitrogen heteroatom situated at position 1 and 4 of the hetero ring system.
The 13C-NMR spectrum of reactive intermediate 7 revealed the presence of nine carbon atoms which include a carbonyl at δ 168.77 ppm, one C=N carbon of imino at δ 140.37 ppm six aromatic carbon at δ 134.91 ppm to 114.52 ppm, and the last carbon close to TMS was CH3 type at δ 20.92 ppm. Careful observation of the signals of 8a in comparison with that of compound 7 showed effective transformation of 2-methyl-4H-3,1-benzoxazin-4-one, 7 to 2,3-dimethyl quinazolin-4(3H)-one, 8a by the virtue of one additional signal at δ 20.92 ppm which was that of second methyl group found in 8a but absent in 7. The most de-shielded signals in the 13C-NMR spectra in all of the compounds varied between d 173.3 and 172.8 ppm which were attributed to the presence of carbonyl of ester in 7 and carbonyl of amide in 8a-q. It was in agreement with the earlier reported value by Al-Kawkabani et al., who investigated the synthesis of novel 2H,8H-pyrano[2,3-f]chromene-2,8-dione based scaffolds under tandem knoevenagel condition24.
Furthermore, the infrared spectrum of the precursor 7 was run in KBr pellet and it showed the presence of C-H aromatic stretching vibrational frequency at 3020 cm-1 while that of C-H aliphatic was noticed as two bands at stretching vibrational frequencies of 2929 cm-1 and 2846 cm-1. The C=O of ester was responsible for the band at 1743 cm-1 which was further confirmed by the presence of C-O of alkoxy group at 1109 cm-1. The absorption bands at ν 1620 cm-1 and 1576 cm-1 depicted the presence of C=C aromatic and C=N imino functionalities respectively. The infrared spectroscopy spectrums of the synthesized compounds were run in KBr pellet using Bruker FT-IR machine. According to the result of the 2-methylsubstituted quinazolinone (8a-q), the C-H band of the aromatic were found in the range 3188 cm-1 (8h) to 3011 cm-1 (8j); while the highest band for C-H of aliphatic was observed in 8b and 8d as 2935 cm-1 and the lowest was at 2845 as seen in compounds 8a and 8e. The bands in the range of 1693 (8g) to 1685 (8i) depicted the presence of C=O of amide in compounds 8a to 8q. The C=C of compounds 8a-q absorbed at stretching vibrational frequency of 1660 cm-1 (8c) to 1600 cm-1 (8l and 8h). The C=N imino functionality was present in all compound 8a-q at the stretching vibrational frequencies at region 1588 cm-1 (as found in 8c) to 1575 cm-1 (which was common to compound 8f, 8l, 8g). It was worthy to note that deformed alkyl group was presence in compound 8a-q, by the virtue of CH3 group in position 2 of the quinazolinone ring and as a result of availability of CH2, CH, in some peculiar compounds. The vibrational frequencies for the CH3, CH2, CH, (deformation) was noticed at 1485 cm-1 highest value of 1485 cm-1 for 8p while the lowest value for this was found at 1370 cm-1 for 8e. The absorption bands at 1248 cm-1 (8m) to 1245 cm-1 (8o and 8n), depicted the presence of C-N functional moieties. Specifically speaking compound 8o alone showed an asymmetry stretching. The frequency herein reported for carbonyl of ester at 1743 cm-1 was further confirmed by comparing it with the findings of Lewis et al. who documented the various C=O stretching absorption vibration frequencies in infrared spectra25 ranged between 1685 cm-1 to 1758 cm-1. The C=N reported herein was within the ranged of the C=N of benzimidazole earlier reported in our previous study2.
The electronic absorption spectra featuring λmax (Log εmax) of the synthesized compounds was run in ethanol because they were all soluble in ethanol and the result is as shown in the experimental section. The lowest wavelength in the electronic transition of the synthesized compounds in 8a-q was found at 206 nm as seen in compounds 8i and 8j respectively. The lowest wavelength for the compounds were as a result of the π→π* transition of C=C which showed the presence of benzene ring in all the compounds. Other transitions found at longer wavelengths were bathochromic in nature and are due to presence of auxochromic group nitrogen atom which is able to delocalize its lone pair into the chromophoric group thereby extending the wavelength and affecting the intensity of the absorbance. The highest wavelength 545 nm (Log εmax = 3.08) was found in the uv electronic transition of the compound 8m. Specifically, the visible absorption spectrum of 8m, for instance, showed a low band at λmax = 221 nm (Log εmax = 5.35) which was the value for the confirmation of presence of phenyl ring while the bathochromic shift observed at λmax = 302 nm (Log εmax = 4.46) was due to the presence of an auxochrome imine (N=C). This was as a result of π→n transition which may be ascribed to the auxochromic C=N group. The noticeable peaks seen at λmax = 545 nm (Log εmax = 3.08) might be due to the π→n transition peculiar to the NH herein present in compound 2m, although, with low log of molar absorptivity. It is interesting to note that all the compounds have electronic transition which culminated into specific peaks in their uv spectra, including even the whitish coloured compounds. In addition, the uv-visible spectra of the quinazoline-4-one, 8a-8q experienced bathochromic shifts of about 15 – 30 nm to give higher max values at above 351 nm, which may be ascribed to the chromophoric C=N group; characteristic of K bands of C=N functional group26.
Conclusion
As envisaged from reviewed literature and chemical behavioural pattern of 2-methyl-4H-3,1-bnezoxazin-4-one, 7, the thermal annelation of this reactive intermediate with various amino-based compounds to afford the corresponding 2-methyl-3-substitutedquinazolin-4(3H)-one derivatives, 8a-q was successful. The reaction optimization study revealed the crucial role of the careful choice of solvent and thermodynamic parameter in order to achieve the targeted compounds in good to excellent yields even in the absence of catalyst. The spectroscopic assignments for structural elucidation was in concordance with the proposed structures for the synthesized compounds. The titled compounds are good candidate for further study in term of the investigation of their antimicrobial and antimalarial potential for further drug development.
Acknowledgment
O.O.A. gratefully acknowledges The World Academy of Sciences for the sponsorship of this project under the TWAS Research Grants Programme in Basic Sciencefor Individual Scientists (Grant No. 14-069 RG/CHE/AF/AC_1).
References
- Ajani, O. O. Eur. J. Med. Chem., 2014, 85, 688-715.
CrossRef - Ajani, O. O., Aderohunmu, D. V., Olorunshola, S. J., Ikpo, C. O., Olanrewaju, I. O. Orient. J. Chem., 2016, 32(1), 109-120.
CrossRef - Pedro, M., João, J., Sofia, S., Luis, R. R., Catarina, R. R., Pedro, V. B., Alexandra, R. F. Molecules, 2015, 20, 16852-16891.
CrossRef - Ajani, O. O., Isaac, J. T., Owoeye, T. F., Akinsiku, A. A. Int. J. Biol. Chem., 2015, 9, 148-177.
CrossRef - Nayaka, R. B., Neeladri, S., Yenumalapadmavathi, Mannem, S. Creat. J. Pharm. Res., 2015, 1, 37-45.
- Vijayakumar, B., Prasanthi, P., Muni T. K., Makesh, K. R. K., Nishanthi, P., Nagendramma, M., Nishanthi, M. IJMCA, 2013, 3(1), 10-21.
- Sharma, G. V. R., Robert, A. R. Int. J. Adv. Pharm., Biol. Chem., 2012, 1(3), 337-341.
- McLaughlin, N.P., Evans, P, Pines. M. The chemistry and biology of febrifugine and halofuginone. Bioorg. Med. Chem., 2014, 2, 1993-2004.
CrossRef - Al-Jaber, N. A., Karah, M. M., Dahlous, K. A., Alafeefy, A. M. Int. J. Chem. Applied Biol. Sci., 2014, 1, 65-72.
CrossRef - Serya, R. A. T., Abbas A. H., Ismail, N. S. M., Esmat, A. and El Ella. Chem. Pharm. Bull., 2015, 63, 102-116.
CrossRef - Zhu, .X, Van Horn, K. S., Barber, M. M., Yang, S., Wang, M. Z., Manetsch, R., Werbovetz, K. A. Bioorg. Med. Chem., 2015, 23, 5182-5189.
- Bule, M. H., Haymete, A., Kefale, B. Drug Design., 2015, 4, 121. 6 pp
- Kumar, S., Mane, B., Siddappa, K. J. Chem., Biol. Phys. Sci., 2015, 5(3), 2802-2805.
- Babu, N. R., Sreenivasulu, N., Yenumalapadmavathi, Srivarsha, M. Creat. J. Pharm. Res., 2015, 1(1), 37-45.
- Wan, Z., Hu, D., Li, P., Xie, D., Gan, H. Molecules, 2015, 20, 11861-11874.
CrossRef - Karnakar, K., Shangkar, J., Murthy, S.N., Ramesh, K., Nageshwar, Y.V.D. Synlett, 2011, 2011, 1089-1096.
- Chen, M., Zhang, M., Xiong, B., Tan, Z., Lv, W., Jiang, H. Org. Lett., 2014, 16, 6028-6031.
CrossRef - Hédou, D., Guillon, R., Lecointe, C., Logé, C., Chosson, E., Besson, T. Tetrahedron, 2013, 69, 3182-3191.
CrossRef - Reddy, G. R., Reddy, T. R., Chary, R. G., Joseph, S. C., Mukherjee, S., Pal. M. Tetrahedron Lett., 2013, 54, 6744-6746.
CrossRef - Zhou, Y. J., Zhang, M. M., Li, Y. L., Liu, Y., Wang, X. S. Tetrahedron, 2014, 70, 3440-3446.
CrossRef - Venkateswarlu, S., Satyanarayana, M., Ravikiran, P., Vijayakumar, A. Tetrahedron Lett., 2013, 54, 4512-4514.
CrossRef - Hassan, G. S., Kadry, H. H., Abou-Seri, S. M., Ali, M. M., Mahmoud, E. E. D. Bioorg. Med. Chem., 2011, 19, 6808-6918.
CrossRef - Ajani, O. O., Obafemi, C. A., Nwinyi, O. C., Akinpelu, D. A. Bioorg. Med. Chem., 2010, 18(1), 214-221.
CrossRef - Al-Kawkabani, A., Boutemeur-Kheddis, B., Makhloufi-Chebli, M., Hamdi, M., Talhi, O., Silva, A.M. S. Tetrahedron Lett., 2013, 54, 5111-5114.
CrossRef - Lewis, R. N., McElhaney, R. N., Pohle, W., Mantsch, H. H. Biophys. J., 1994, 67(6), 2367-2375.
CrossRef - Komurcu, S. G., Rollas, S., Uglen, M., Gorrod, J.W. Boll. Chim. Farmac., 1995, 134, 375-379.
Accepted on: March 29, 2017







ISSN Online: 2231-5039

![]()




{kind=link}