Headspace-Single Drop Microextraction Followed by Gas Chromatographic Determination of Key Aroma Compounds in Tomato Fruits and Their Sample Products
Department of Chemistry and Center for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand.
Corresponding Author E-mail: sakcha2@kku.ac.th
DOI : http://dx.doi.org/10.13005/ojc/320301
Download this article as:
![]()
A headspace-single drop microextraction (HS-SDME) is a good choice to analyze volatile and semivolatile compounds in different matrices without any interference of the sample matrix. HS-SDME spends a very little solvent consumption for the determination of volatile aroma compounds (VACs). In this study, some VACs including 2-methylbutyraldehyde, 3-methyl-1-butanol, sec-butyl acetate, 1-octen-3-one and trans,trans-2,4-decadienal were performed with hexadecane as extraction solvent. The parameters affecting the HS-SDME method were investigated in details including extraction temperature, enrichment time, extraction time and sample volume. The optimum conditions were consisted of 45oC extraction temperature, 15min enrichment, 20min extraction time and 3mL sample volume. Comparison between liquid-liquid extraction (LLE) and HS-SDME for quantitative analysis was carried out using tert-butanol as an internal standard. From results revealed that the recoveries were obtained between 81.06-90.55% for LLE and 80.26-90.09% for HS-SDME. The results demonstrated that the proposed method could be successfully applied for the determination of VACs.
KEYWORDS:Headspace-single drop microextraction; Gas chromatography; Volatile aroma compounds; Tomato
Introduction
Tomato (Lycopersicon esculentum Mill.), a member of the Solanaceae family, is one of the most important sources of essential nutrients worldwide. Botanically, it is a berry fruit, but it is cultivated and used worldwide as a vegetable, being the third in the vegetables and the tenth biggest economic production. Tomato is one of the most important vegetables in the world, with the total production of 145.8 million tonnes recorded in 2010 and 161.79 million tonnes in 20121.This huge demand for tomato is certainly due to its classification as functional food that in turn arises from the association between tomato consumption and reduced likelihood of certain types of cancers and cardiovascular diseases2.
The aroma compositions of fresh tomato have been studied and more than 400 volatile compounds have been identified3. The volatile aroma compounds (VACs) present in fresh and processed tomato are included in various chemical classes such as ketones, aldehydes, alcohols, phenols, esters, ethers, hydrocarbons, sulfur compounds, nitrogen-containing compounds, furan, free acids and lactones4-11. The most important volatile compounds are thought to be cis-3-hexenal, β-ionone, hexanal, β-damascenone, l-penten-3-one, 2+3-methylbutanol, trans-2-hexenal, cis-3-hexenol, 2-isobutylthiazole, l-nitro-2-phenylethane, trans-2-heptenal, phenylacetaldehyde, methylsalicylate, 3-methylbutanal, 2-phenylethanol, 6-methyl-5-hepten-2-one, and geranylacetone12-14. In addition, furaneol15, hexanol, methional and l-octen-3-one16 may also contribute to the tomato aroma taste.
Moreover, previous research reported that 2-butylacetate, 3-methyl-1-butenol, 2-methylbutanal, 1-octen-3-one, and trans,trans-2,4-decadienal define aroma intensity within tomato. In addition, five volatile aroma compounds were identified as chemical markers of tomato and were proved to be important contributors to sweetness of tomato17. Therefore, the researcher was interested in five volatile aroma compounds as target analytes.
Various methods of sample preparation for the volatile aroma compounds including solvent extraction or liquid-liquid extraction (LLE)18, solid phase extraction (SPE)19,direct immersion (DI) and headspace (HS)-solid phase microextraction (SPME) have been recently applied to their determination20-22. Apart from SPME, more attention has been focused on miniaturized and solvent-free liquid-phase extraction technique. Liquid phase microextraction (LPME) which is also known as a single-drop microextraction (SDME) is included. The SDME, a sample preparation technique introduced since 1996, has attracted increasing attentions23-25. It is commonly combined with gas chromatography-flame ionization detector (GC-FID). It is now becoming one of the most common methods of sample preparation, particularly for the extraction of organic compounds from environmental and biological samples26-28. Moreover, HS-SDME is a good choice to analyze volatile and semi-volatile compounds in different matrices without any interference of the sample matrix29,30. To date, it has become a very popular liquid phase microextraction technique. This extraction method is quick, easy to operate, and inexpensive. Since very little solvent is used, it is considered an environment friendly approach. In addition, renewable extraction phase in SDME which eliminates possible memory effects as fresh solvent-drop is used for each extraction31. The HS-SDME is a well-designed method that is able to overcome some of the drawbacks of SPME, as a wide variety of solvents and trapping agents can be used. Hence, it has been successfully applied in various fields. In this method, a single drop of an extractant of a few micro-scale volume suspended to the tip of a microsyringe. HS-SDME in which the extractant droplet is held above the sample31. After extraction of volatile organic compounds, the microdrop is retracted into the microsyringe and injected directly into GC for further analysis. This novel technique eliminates disadvantages of the conventional LLE. Since very little solvent is used, there is minimal exposure to the toxic organic solvents for the operator. One advantage of SDME is the integration of extraction, concentration and sample introduction into a single step25,33,34. Presently, HS-SDME has been successfully used for the extraction of volatile to semi-volatile compounds, without any interference from the sample matrices. Until now, no data is available for the five target analytes by HS-SDME. Therefore, the aim of this study was to optimize and validate the HS-SDME method in association with GC for determining five volatile aroma compounds as chemical markers in tomato fruits and their products.
Materials and Methods
Tomato samples
Tomato juices such as Brook, Campbell’s, Mica, UFC, Kagome were purchased from Tops supermarket, Central Plaza Khon Kaen. Malee fruit juice was obtained from Tesco Lotus department store, Khon Kaen. Doikham fruit juice was bought from 7-Eleven shop, Khon Kaen. Tomato paste samples including Mica, Mutti and Pomi were also purchased from Tops supermarket, and KFC tomato paste was obtained from KFC shop, Khon Kaen. For tomato fresh fruits, Holland tomato, orange cherry tomato, red cocktail tomato, and strawberry tomato were also bought from Tops supermarket, whereas Cherry tomato and Sida tomato were obtained from Tesco Lotus.
Chemicals and reagents
The standards 2-methylbutyraldehyde (b.p.90.0-92.0 oC), sec-butyl acetate (b.p.111.0-112.0 oC), 1-octen-3-one (b.p.174.0-182.0 oC), trans,trans-2,4-decadienal (b.p.114.0-116.0 oC) were purchased from Aldrich-Chemistry (USA) and 3-methyl-1-butanol (b.p.130.0 oC) was purchased from Sigma-Aldrich (USA). tert-Butanol (b.p.81.7-82.7 oC) as an internal standard (IS) was purchased from Carlo Erba (France). The chemical structure of VACs standards is shown in Fig. 1. The stock solutions of each volatile aroma compounds were prepared by dissolving in dichloromethane (RCI Labscan, Thailand) and kept in the dark at 4 oC. Single drop solvents namely, 1-octanol (b.p.196.0 oC) and hexadecane (b.p.287.0 oC) were also purchased from Sigma-Aldrich (USA).
Preparation of standard volatile aroma compounds (VACs)
The stock standards of each of volatile aroma compounds (2-methylbutyraldehyde, 3-methyl-1-butanol, sec-butyl acetate, 1-octen-3-one and trans,trans-2,4-decadienal) were accurately prepared by dissolving in dichloromethane and stored at 4oC. Their working standard solutions of lower concentrations were daily prepared by an appropriate step dilution in the same solvent.
Instrumentation
Single drop microextraction was performed in a 10 mL glass vial (2.0×4.5 cm, Thailand) that tightly closed with septum inside the cap. The glass vial with a small magnetic bar (10.0×6.0 mm, Union science, Thailand) was placed in a beaker (100 mL, Pyrex, USA) with a big magnetic bar (30.0×7.0 mm, Union science, Thailand), which placed on a magnetic stirring hotplate (MR 3001, Heidolph, Germany) maintaining at a desirable temperature and stirring rate.
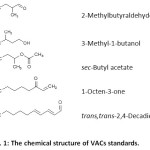 |
Figure 1: The chemical structure of VACs standards. Click here to View Figure |
The volatile aroma compounds in various tomato samples were analyzed by GC-FID (Trace GC, Thermo Finnigan, Italy). A capillary column DB-5, 30.0 m, 250 µm ID, 0.25 µm film thickness was employed. Helium was used as carrier gas at a flow rate of 1 mL min-1. The injector temperature was set at 260 oC. The injection volume was 1 µL and all injections were made in split mode (split ratio, 50:1). The detector temperature was maintained at 300 oC.
Optimization of GC conditions
The optimum chromatographic separation of the five target VACs including an internal standard used was studied by simply varying the temperature gradient program of the GC oven.
Optimization of HS-SDME
Various parameters in HS-SDME affecting to the extraction efficiency, including the extraction solvent, extraction temperature, enrichment time, extraction time and sample volume were optimized in details.
HS-SDME procedure
Sample solution was added into a 10 mL septum sealed vial and small magnetic stir bar was introduced in the vial. Then, the vial was placed in 100 mL beaker with bigger magnetic stir bar and water. It was placed on magnetic stirring hotplate previously programmed at a desirable temperature and was maintained under magnetic stirring at 700 rpm. The analytes was transferred from solution to the headspace phase due to the gas-liquid distribution equilibrium at which this procedure was “extraction step” and the stirring time of this period was called “extraction time”. After extraction step, the stirrer was turned off. Then, a 1 µL single drop solvent containing 100 µg mL-1 of tert-butanol used as an internal standard was withdrawn into a microsyringe, the microsyringe needle was passed through the vial septum and the end of needle was located about 1 cm above the surface of the solution. Mass transfer of analytes from gas phase to the suspended single drop solvent continued until the enrichment was completed, indicating “enrichment step”. The time lag of this period was so-called the “enrichment time”. After the enrichment, the extract was withdrawn into the microsyringe, the microsyringe was removed from the vial and the extract was injected into the GC for analysis.
Real sample analysis
Tomato sample (3 mL for tomato juice and 3 g for tomato paste and tomato fruit) was placed into conical centrifuge tube and firstly extracted by 2 mL dichloromethane. The mixture portion was thoroughly vortexed for 5 min, and then centrifuged for 10 min. The extracted solution was removed using a syringe (5 mL, Nipro, Thailand) and injected into 5 mL volumetric flask for dilution in dichloromethane. Then a 3 mL mixture solution was added into 10 mL glass vial. The solution was stirred with a magnetic stir bar for 20 min. In HS-SDME, using GC syringe (10 µL, Hamilton, USA) was fixed through its septum of the headspace vial, and the needle tip appeared 1 cm over the surface of solution. A 1 µL suspended microdrop of hexadecane containing 100 mg mL-1 of tert-butanol (as IS) was withdrawn into a microsyringe and then was suspended at the needle tip of microsyringe for 15 min. After extraction, 1.0 µL of the resulting extract was retracted into the microsyringe and was immediately injected into the GC for analysis.
For liquid-liquid extraction (LLE), tomato sample (3 mL for tomato juice and 3 g for tomato paste and tomato fruit) was placed into conical centrifuge tube and extracted by 2 mL dichloromethane. The mixture portion was thoroughly vortexed for 5 min, and then centrifuged for 10 min. The clear extract was removed using a syringe (3 mL, Nipro, Thailand) and injected into 5 mL volumetric flask for dilution in dichloromethane. Then, 1.0 µL of the resulting extract was injected into the GC.
The recovery study
To focus on an accuracy of the method, the recovery study was carried out by spiking of known concentrations (3-10 µg mL–1) of the mixed VACs standard into the samples prior to sample pretreatment and extraction by LLE and HS-SDME. All experiments were performed with five replicates.
Results and Discussion
Optimization of GC conditions
The oven temperature program started at 40 oC with 1 min holding time, then increased up to 220 oC with a ramp rate of 10 oC min-1 and held at this temperature for 1 min.
Chromatographic separation and analytical performance
Several temperature programs were tested in order to obtain the best separation of target volatile compounds in shortest analysis time. Under the adequate GC conditions, separation of five VACs was achieved within 20 min, with the following order of the separation: 2-methylbutyraldehyde (tR 4.75 min), 3-methyl-1-butanol (tR 5.72 min), sec-butyl acetate (tR 6.04 min), tert-butanol (as IS) (tR 6.16 min), 1-octen-3-one (tR 9.98 min), and trans,trans-2,4-decadienal (tR 14.73 min).
Optimization of HS-SDME
To achieve highest extraction performance, the experimental parameters including extraction solvent, extraction temperature, enrichment time, extraction time and sample volume were optimized. When one parameter was changed, other parameters were fixed at their optimal values.
Effect of the type of extraction solvent
The suspended extraction solvents were the first step in optimization procedure of HS-SDME. The characteristic requirements of the suspended solvent in HS-SDME are good chromatographic behavior, high boiling point, low vapor pressure and low volatility in order to minimize any evaporation during the extraction process, rather high viscosity, polarity similar to VACs, and low toxicity. Both 1-octanol and hexadecane were chosen to study in order to select the best solvent for the extraction of the VACs from tomato and their fruit products.
Figure 2 presents the maximum peak area of target VACs which was obtained by using hexadecane as the single drop extraction solvent. In fact, the suspended solvents used were different in their polarity and boiling point. Hexadecane is a very stable suspended
solvent because it has higher boiling point than 1-octanol. In addition, the non-polar long chain hydrocarbon of hexadecane was reasonably satisfied to be the extraction solvent for all hydrophobic VACs by HS-SDME. By comparing between the two solvents, it was shown that hexadecane resulted in a more efficient extraction of VACs. Thus, 1 µL of hexadecane containing 100 µg mL-1 tert-butanol was chosen as the extraction solvent for further study.
Effect of extraction temperature
For volatile analytes, the total extraction efficiency is described which is a compromise between mass transfer of the analytes from the sample solution to the headspace and then from the headspace to the extraction solvent. Higher temperatures lead to higher vapor pressure of the analyte and hence its concentration in headspace increase35. The experimental results show that the extraction efficiency increased as an increasing of temperature, (Fig. 3).
 |
Figure 2: Effect of hexadecane and 1-octanol as the extraction solvents on the peak area of the VACs. Other experimental conditions: 100 µg mL-1 VACs concentration; 3 mL sample volume; 40oC extraction temperature; 20 min extraction time; 10 min enrichment time.
|
This reveals that at higher temperatures, the vapor pressure of the analytes and their concentrations in the headspace increase. The amounts of the extracted analytes decrease above 45 oC, probably due to the decreasing of the partition coefficients of analytes between headspace and the extraction phase. Conversely, high temperatures are due to the solvent drop damage and decrease the reproducibility of the extraction procedure36,37. In this study, it was found that if the extraction temperature is higher than 49 oC, the suspended microdrop is rather unstable. Thus, the extraction temperature of 45 oC was the most suitable conditions.
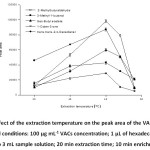 |
Figure 3: Effect of the extraction temperature on the peak area of the VACs. Other experimental conditions: 100 µg mL-1 VACs concentration; 1 µL of hexadecane drop was exposed to 3 mL sample solution; 20 min extraction time; 10 min enrichment time. |
Effect of enrichment time
An enrichment time is the exposure time of the suspended extraction solvent in the headspace. The amount of the target analytes will be saturated in the microdrop until their dynamic equilibrium state is established. The effect of enrichment time was studied in the range of 1-20 min. From Fig. 4, it is shown that the longer the enrichment time, the lower stability of the suspended microdrop, leading to low extraction efficiency of VACs. However, the peak areas of target analytes firstly increase with increasing enrichment time and then decrease when the time is no longer than 15 min. The reason may be accounted by the back-extraction from the microdrop into the headspace. The HS-SDME is not an exhaustive extraction method, and the extraction efficiency does not always increase with the increase of enrichment time38. The analytes are distributed among the sample solution phase, the headspace and the suspended microdrop. Hence, the amount of the analyte transferred into the suspended microdrop reaches its maximum when the dynamic equilibrium state is established39.As soon as the concentration of the analytes in headspace is lower than the equilibrium value, the analyte molecules begin to diffuse from the suspended microdrop into the gas phase. The experiment did not conduct after 20 min because of loss of the extraction solvent volume at longer times. Therefore, an enrichment time of 15 min was chosen for the present work.
 |
Figure 4: Effect of the enrichment time on the peak area of the VACs. Other experimental conditions: 100 µg mL-1 VACs concentration; 1 µL of hexadecane drop was exposed to 3 mL sample solution; 45oC extraction temperature; 20 min extraction time. |
Effect of extraction time
Extraction time is the time for agitation of the sample solution by magnetic stir bar. The effect of extraction time at a range of 5 to 40 min was investigated and the experimental results are shown in Fig. 5. It was found that the peak area of these VACs increase with the increase of the extraction time from 5 to 20 min. Beyond 20 min the peak area decreased. This is due to the decomposition and back-extraction of analytes caused the by overlong extraction time. The result shows that extraction time for 20 min, peak area of all analytes are the highest40. Thus, compromise time of 20 min was chosen.
 |
Figure 5: Effect of the extraction time on the peak area of the VACs. Other experimental conditions: 100 µg mL-1 VACs concentration; 1 µL of hexadecane drop was exposed to 3 mL sample solution; 45oC extraction temperature; 15 min enrichment time. |
Effect of sample volume
Sample volume relates directly to the magnitude of the headspace, and may relate to an extraction efficiency. The effect of sample volume on the extraction efficiency was studied in range of 1-7 mL. According to this observation, the highest peak area occurred at 3 mL (Fig. 6). By increasing the sample volume up to 3 mL, the extraction efficiency increased and the peak area of the target analytes decreased above 3 mL with some fluctuation between 5 mL and 7 mL. This phenomenon could be attributed to the fact that an increasing of the sample volume can lead to the decrease of the headspace volume, which accelerates the diffusion of the analytes into the suspended solvent until they reach their equilibrium saturation. Under fixed stirring speed with a large volume, the convection is not as good in the sample solution, resulting in less extraction. Thus, 3 mL of the sample volume was chosen.
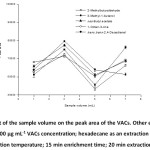 |
Figure 6: Effect of the sample volume on the peak area of the VACs. Other experimental conditions: 100 µg mL-1 VACs concentration; hexadecane as an extraction solvent; 45oC extraction temperature; 15 min enrichment time; 20 min extraction time. |
Method validation
To evaluate the optimized extraction conditions of the HS-SDME method, the dynamic linear range, limit of detection (LOD), limit of quantification (LOQ), accuracy and precision were investigated, and comparison of the HS-SDME with LLE method were also carried out41,42.
The precision of the proposed method was presented as the relative standard deviations of the peak area. The intra-day precision was deduced from five replicates in one day (n=5) and inter-day precision was calculated from the experiments carried out in 3 days (n=3×5).
Analytical figures of merit
The calibration curves were made by plotting peak area ratios versus the concentrations of the analytes under the optimum conditions. The dynamic linear range, correlation coefficients, the LOD and LOQ, based on signal to noise ratio of 3 and 10, respectively, were obtained. Both LOD and LOQ were performed with five replicates and were calculated by using peak height. The analytical results of the validated methods for both HS-SDME and GC-FID are shown in Table 1.
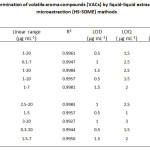 |
Table 1: Analytical performance for determination of volatile aroma compounds (VACs) by liquid-liquid extraction (LLE) and the headspace single drop microextraction (HS-SDME) methods
|
Application to real samples
The developed HS-SDME procedure has been applied for the determination of target VACs in numerous different tomato juices, tomato paste and tomato fruits. The comparison between LLE method and HS-SDME method for quantitative analysis was carried out using tert-butanol as an internal standard as shown in Table 2. It was found that the contents of VACs in tomato samples determined by HS-SDME were higher than those of LLE. It is, thus, indicated that the HS-SDME has also been developed for the preconcentration of VACs in tomato samples followed by GC analysis. 2-Methylbutyraldehyde and 1-octen-3-one were only found in tomato juices studied. It is indicated that the two volatile compounds are possibly occurred during a production process. Furthermore, trans,trans-2,4-decadienal was found in some tomato samples whereas 3-methyl-1-butanol was found in all samples studied. More broadly, the results suggest that 3-methyl-1-butanol is considered as chemical marker, so it can be sensed for tomato’s aroma volatile odor in the processed foods. The method recoveries were found in the ranges of 81.1-90.6% and 80.3-90.1% for LLE and HS-SDME, respectively (Table 3). The obtained results support that the proposed method has been proven to be suitable for the determination of the VACs in tomato samples.
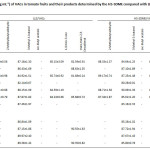 |
Table 2: The contents (µg mL-1) of VACs in tomato fruits and their products determined by the HS-SDME compared with LLE (Avg. ±SD, n = 5) (cont.)
|
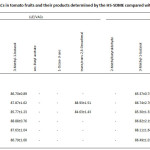 |
Table 3: The recovery (%) for VACs in tomato fruits and their products determined by the HS-SDME compared with LLE (Avg. ±%RSD, n = 5) (cont.)
|
Conclusion
In the present study, the results revealed that the dynamic linear ranges were found to be 0.1-20 µg mL-1 and 0.3-20 µg mL-1 for LLE and HS-SDME, respectively. The LODs were ranged from 0.5-1.5 µg mL-1 for LLE and 0.5-1.5 µg mL-1 for HS-SDME. The LOQs were in the ranges of 1.5-2.5 and 1.0-3.0 μg mL–1 for LLE and HS-SDME, respectively. The intra-day RSDs of peak area were less than 3.23 and 2.95% for LLE and HS-SDME, respectively. For inter-day experiments, the RSD values were below 4.66% (LLE) and 3.39% (HS-SDME) in terms of peak area. The applicability of the developed method was finally examined for the determination of the target volatile aroma compounds in different tomato samples. The results indicated that 3-methyl-1-butanol could be considered as a chemical marker for this case study. The method recoveries were obtained in the ranges of 81.06-90.55 % and 80.26-90.09 % for LLE and HS-SDME, respectively, satisfactorily subjecting for their dynamic extraction efficiency. The advantages of the proposed method gain much more benefits as quick, easy to operate and inexpensive procedure. In addition, the microextraction process consumes little volume of solvent, so it is considered as an environmentally friendly approach for green chemistry. Good analytical performances of the method have been successfully obtained for the determination of VACs in various tomato samples.
Acknowledgements
This research was financially supported by the Higher Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, through the Food and Functional Food research Cluster of Khon Kaen University, and the Center for Innovation in Chemistry (PERCH-CIC), Commission on Higher Education, Ministry of Education are gratefully acknowledged.
References
- FAOSTAT, Food and Agriculture Organization of the United Nations Statistical Databases (2012) FAOSTAT. http://faostat.fao.org. Accessed 14 June 2012
- Giovannetti, M.; Avio, L.; Barale, R.; Ceccarelli, N.; Cristofani, R.; Iezzi, A.; Mignolli, F.; Picciarelli, P.; Pinto, B.; Reali, D.; Sbrana, C.; Scarpato, R. Br. J. Nutr. 2012, 107, 242-251
CrossRef - Petrὸ-Turza, M. Food Rev. Int. 1987, 2, 309-351
CrossRef - Baldwin, E.A.; Nisperos-Carriedo, M.O.; Baker, R.; Scott, J.W. J. Agric. Food Chem. 1991, 39, 1135-1140
CrossRef - Buttery, R.G.; Teranishi, R.; Ling, L.C. J. Agric. Food Chem. 1987, 35, 540-544
CrossRef - Buttery, R.G.; Takeoka, G.; Teranishi, R.; Ling, L.C. J. Agric. Food Chem. 1990, 38, 2050-2053
CrossRef - Buttery, R.G.; Teranishi, R.; Flath, R.A.; Ling, L.C. J. Agric. Food Chem. 1990, 38, 792-795
CrossRef - Linforth, R.S.T.; Savary. I.; Pattenden. B.; Taylor. A.J. J. Sci. Food Agric. 1994, 65, 241-247
CrossRef - Petrὸ-Turza, M. Food Rev. Int. 1986, 2, 309-351
CrossRef - Riley, J.C.; Willemot, C.; Thompson, J.E. Postharvest Biol. Technol. 1996, 7, 97-107
CrossRef - Servili, M.; Selvaggini, R.; Taticchi, A.; Begliomini, A.L.; Montodoro, G.F. Food chem. 2000, 71, 407-415
CrossRef - Krumbein, A.; Auerswald, H. Nahrung 1998, 42, 395-399
CrossRef - Baldwin, EA.; Scott, JW.; Einstein, M.A.; Malundo, T.M.M.: Carr, BT.; Shewfelt, R.L. J. Am. Soc. Hortic. Sci. 1998, 123, 906-915
- Tandon, K.S.; Baldwin, E.A.; Shewfelt, R.L. Postharvest Biol. Technol. 2000, 20, 261-268
CrossRef - Buttery, R.G.; Takeoka, G.R.; Ling, L.C. J. Agric. Food Chem. 1995, 43, 1638-1640
CrossRef - Tandon, K.S.; Jordán, M.; Goodner, K.L.; Baldwin, E.A. Proc. Fla. State Hort. Soc. 2001, 114, 142-144
- Tieman, D.; Bliss, P.; McIntyre, L.M.; Blandon-Ubeda, A.; Bies, D.; Odabasi, A.Z.; Rodríguez, G.R.; Knaap, E.V.D.; Taylor, M.G.; Goulet, C.; Mageroy, M.H.D.; Snyder, J.; Colquhoun, T.; Moskowitz, H.; Clark, D.G.; Sims, C.; Bartoshuk, L.; Klee, H.J. Current Biology 2012, 22, 1035-1039
CrossRef - Mamede, M.E.O.; Pastore, G.M. Food chem. 2006, 96, 586-590
CrossRef - Ahrer, W.; Scherwenk, E.; Buchberger, W. J. Chromatogr. A 2001, 910, 69-78
CrossRef - Figueira, J.; Camara, H.; Pereira, J.; Camara, J.S. Food Chem. 2014, 145, 653-663
CrossRef - Feudo, GL.; Macchione, B.; Naccarato, A.; Sindona, G.; Tagarelli, A. Food Res. Int. 2011, 44, 781-788
CrossRef - Zanjani, M.K.; Yamini, Y.; Shariati, S. J. Hazard. Mater. 2006, 136, 714-720
CrossRef - Jeannot, M.A.; Cantwell, F.F. Anal. Chem. 1997, 69, 235-239
CrossRef - Jeannot, M.A.; Cantwell, F.F. Anal. Chem. 1996, 68, 2236-2240
CrossRef - He, Y.; Lee, H.K. Anal. Chem.1997, 69, 4634-4640
CrossRef - Patel, K.; Mehta, P.; Sahoo, U.; Sen, B.D. Int. J. Chem. Tech. Res. 2010, 2, 1638-1652
- He, X.; Zhang, F.; Jiang, Y, J. Chromatogr. Sci. 2012, 50, 457-463
CrossRef - Riccio, D.; Wood, D.C.; Miller, J.M. J. Chem. Educ. 2008, 85, 965-968
CrossRef - Theis, A.L.; Waldack, A.J.; Hansen, S.M.; Jeannot, M.A. Anal. Chem. 2001, 73, 5651-5654
CrossRef - Shen, G.; Lee, H.K. Anal. Chem. 2003, 75, 98-103
CrossRef - Hakkarainen, M. J. Biochem. Biophys. Methods 2007, 70, 229-233
CrossRef - Pena-Pereira, F.; Lavilla, I.; Bendicho, C. Anal. Chim. Acta. 2009, 631, 223-228
CrossRef - Mohammadi, A.; Alizadeh, N.; J. Chromatogr. A. 2006, 1107, 19-28
CrossRef - Shariati-Feizabadi, S.; Yamini, Y.; Bahramifar, N. Anal. Chim. Acta. 2003, 489, 21-31
CrossRef - Wu, Y.; Xia, L.; Chen, R.; Hu, B. Talanta 2008, 74, 470-477
CrossRef - Wang, L.; Li, D.; Bao, Ch.L.; You, J.Y.; Wang, Z.M.; Shi, Y.H.; Zhang, H.Q. Ultrason.Sonochem 2008, 15, 738-746
CrossRef - Wang, Z.M.; Ding, L.; Li, T.C.; Zhou, X.; Wang, L.; Zhang, H.Q.; Liu, L.; Li, Y.; Liu, Z.H.; Wang, H.J.; Zeng, H.; He, H. J. Chromatogr. A 2006, 1102, 11-17
CrossRef - Jalali, H.M.; Sereshti, H. J. Chromatogr. A 2007, 1160, 81-89
CrossRef - Besharati-Seidani, A.; Jabbari, A.; Yamini, Y. Anal. Chim. Acta 2005, 530, 155-161
CrossRef - Wang, L.; Wang, Z.; Zhang, H.; Li, X.; Zhang, H. Anal. Chim. Acta 2009, 647, 72-77
CrossRef - Omar, M.N.; Muhd Nor; N.M.; Idris. N.A. Oriental J. Chem., 2009, 25(4), 825-832.
- Montazeri, N.; Pourshamsian, K.; Barami, Z.; Alidorrieh, S. Oriental J. Chem., 2011, 27(4): 1317-1324.
Accepted on: June 11, 2016







ISSN Online: 2231-5039

![]()




{kind=link}