Design and synthesis of two alkaloids derivatives using some chemical tools
DOI : http://dx.doi.org/10.13005/ojc/31.Special-Issue1.11
Download this article as:
![]()
Several alkaloids derivatives have developed; nevertheless, expensive reagents and special conditions are required. Therefore, in this study, two alkaloids (brucine and strychnine derivatives) were prepared using several strategies; the structure of compounds obtained was confirmed by elemental analysis, spectroscopy and spectrometry data. In conclusion, the methods used offers some advantages such as good yields, simple procedure, low cost, and ease of workup.
KEYWORDS:Synthesis; Brucine; Strychnine
Introduction
Since several years ago, several alkaloids have been prepared for its use in different biological and analytical methods1-3. For example, a study showed the synthesis of N-chloromethylbrucine chloride by the reaction of brucine with dichloromethane4. Other data have shown the preparation of a brucine derivative (brucidine) by electrolytic reduction of brucine5. Additionally, there are reports of the synthesis of N-(5-carboxypentyl)brucinium bromide via N-alkylation of brucine with 6-bromohexanoic acid6. Other study showed the preparation of brucinium hydrogen (S)-malate pentahydrate and anhydrous brucinium hydrogen (2R,3R)-tartrate by the reaction between brucine and D-L-malic acid or L-tartaric acid in ethanol/water7. Additionally, the porphyrin-brucine conjugate was synthetized by the N-alkylation of brucine with alkylbromotetraphenylporphyrin derivatives8. Recently, a brucine derivative (N1-(2,3-dimethoxy strychnidin-10-yliden)-ethane-1,2-diamine) was synthetized by the reaction of brucine and ethylenediamine using boric acid as catalyst9. In addition, other study showed the synthesis of a brucine-dihydropirymidine derivative using the multi-component system (brucine, benzaldehyde and thiourea)10. All these experimental data show several procedures for synthesis of brucine derivatives; nevertheless, expensive reagents and special conditions are required. Therefore, in this study two new alkaloids derivatives were synthetized using some chemical tools.
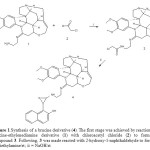 |
Figure 1: Synthesis of a brucine derivative (4). The first stage was achieved by reaction of a brucine-ethylenediamine derivative (1) with chloroacetyl chloride (2) to form the compound 3. Following, 3 was made reacted with 2-hydroxy-1-naphthaldehyde to form 4.i = triethylamine/rt; ii = NaOH/rt Click here to View figure |
Experimental
The brucine-ethylenediamine derivative(N1-(2,3-dimethoxy strychnidin-10-yliden)-ethane-1,2-diamine) was prepared according to previously reported method9. The other compounds evaluated in this study were purchased from Sigma-Aldrich Co. Ltd. The melting points for the different compounds were determined on an Electrothermal (900 model). Infrared spectra (IR) were recorded using KBr pellets on a Perkin Elmer Lambda 40 spectrometer. 1H and 13C NMR spectra were recorded on a Varian VXR-300/5 FT NMR spectrometer at 300 and 75.4 MHz in CDCl3 using TMS as internal standard. EIMS spectra were obtained with a Finnigan Trace GCPolaris Q. spectrometer. Elementary analysis data were acquired from a Perkin Elmer Ser. II CHNS/0 2400 elemental analyzer.
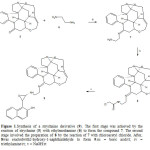 |
Figure 1: Synthesis of a strychnine derivative (9). The first stage was achieved by the reaction of strychnine (5) with ethylenediamine (6) to form the compound 7. The second stage involved the preparation of 8 by the reaction of 7 with chloroacetyl chloride. After, 8was reactedwith2-hydroxy-1-naphthaldehyde to form 9.iii = boric acid/rt; iv = triethylamine/rt; v = NaOH/rt Click here to View figure |
Synthesis of 2-chloro-N-(2-(((4aR,4a1R,5aS,8aR,E)-10,11-dimethoxy-2,4a,4a1,5,5a,7,8, 8a1,15,15a,-decahydro-14H-4,6-methanoindolo[3,2,1-ij]oxepino[2,3,4-de]pyrrolo[2,3-h] quinolin-14-ylidene)amino)ethyl)acetamide (3)
A solution of 1 (200 mg, 0.46 mmol), triethylamine (100 μl, 1.50 mmol) and chloroacetyl chloride (128 μl, 1.60 mmol) in 5 ml of methanol was stirring for 48 h to room temperature. The reaction mixture was evaporated to dryness under reduced pressure. After, the residue was purified by crystallization from methanol:water (2:1) yielding 66 % of product, m.p. 170-172oC; IR (Vmax, cm-1): 3320, 1730 and 1680; 1H NMR (300 MHz, CDCl3) δH: 1.40-1.88 (m, 3H), 2.24-2.34 (m, 2H), 2.70-2.94 (m, 4H), 3.10-3.22 (m, 2H), 3.40 (t, 2H, J = 6.54), 3.55-3.62 (m, 3H), 3.64 (t, 2H, J = 6.54), 3.70 (m, 2H), 3.80 (s, 3H), 3.90 (s, 3H), 4.00 (s, 2H), 4.68 (m, 2H), 5.80-5.86 (m, 2H), 7.00 (broad, 1H), 7.50 (m, 1H) ppm.13C NMR (75.4 Hz, CDCl3) δC:26.60(C-17), 28.05(C-7), 29.20(C-18), 35.04(C-27), 39.90(C-14), 42.38(C-35), 45.60 (C-15), 45.86 (C-3), 50.58 (C-11), 51.32 (C-26), 52.16(C-13), 56.00(C-34), 56.54(C-32), 59.90(C-16), 64.54(C-8), 65.00(C-4), 79.24(C-2), 98.43(C-20), 105.70(C-23), 127.80(C-9), 129.00(C-24), 139.12(C-19), 140.30(C-10), 143.24 (C-22), 147.70(C-21), 147.78(C-6), 162.56(C-9) ppm.EI-MS m/z: 512.21 (M+ 11). Anal. Calcd. for C27H33ClN4O4: C, 63.21; H, 6.48; Cl, 6.91; N, 10.92; O, 12.47. Found: C, 63.16; H, 6.42.
Synthesis of 1-((2S)-3-((2-(((4aR,4a1R,5aS,E)-10,11-dimethoxy-2,4a,4a1,5,5a,7,8,8a1,15, 15a-decahydro-14H-4,6-methanoindolo[3,2,1-ij]oxepino[2,3,4-de]pyrrolo[2,3-h]quino lin-14-ylidene)amino)ethyl)amino)oxiran-2-yl)naphtalen-2-ol (4)
A solution of 3 (200 mg, 0.39mmol), 2-hydroxy-1-naphthaldehyde (70 mg, 0.40mmol), sodium hydroxide (15 mg, 0.37mmol) in 5 ml of ethanol was stirring for 48 h to room temperature. The reaction mixture was evaporated to dryness under reduced pressure. After, the residue was purified by crystallization from methanol:water (4:1) yielding 40 % of product,m.p. 208-210oC; IR (Vmax, cm-1): 3400, 3360, 3324 and 1732; 1H NMR (300 MHz, CDCl3) δH: 1.40-1.80 (m, 3H), 2.20-2.30 (m, 2H), 2.70-2.94 (m, 4H), 2.98-3.00 (m, 2H), 3.10-3.60 (m, 5H), 3.64 (t, 2H, J = 6.44), 3.66 (m, 2H), 3.80 (s, 3H), 3.90 (s, 3H), 4.06 (broad, 2H), 4.24-4.34 (m, 2H), 4.70 (m. 1H), 5.80-5.86 (m, 2H), 7.12-7.40 (m, 4H), 7.52 (m, 1H), 7.66-7.70 (m, 2H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 26.60(C-17), 28.10 (C-7), 29.26(C-18), 39.90 (C-14), 45.58 (C-15), 45.82 (C-3), 47.29 (C-27), 49.50 (C-26), 50.67 (C-11), 52.24 (C-13), 55.89 (C-35), 56.60 (C-33), 59.30 (C-31), 59.86 (C-16), 64.60 (C-8), 65.00 (C-4), 65.48 (C-29), 79.27 (C-2), 98.43 (C-20), 105.70 (C-23), 1.16.88 (C-36), 120.90 (C-38), 122.85 (C-42), 123.00 (C-44), 123.18 (C-41), 126.56 (C-43), 127.86(C-9), 128.92 (C-45), 129.00 (C-24), 130.10 (C-39), 132.66 (C-40), 139.12(C-19), 140.30 (C-10), 143.18 (C-22), 147.70 (C-21), 147.76 (C-6), 152.40 (C-37) ppm.EI-MS m/z: 620.29 (M+ 10). Anal. Calcd. for C37H40N4O5: C, 71.59; H, 6.50; N, 9.03; O, 12.89. Found: C, 71.50; H, 6.46.
Synthesis of 2-(((4aR,4a1R,5aS,8aR,E)-2,4a,4a1,5,5a,7,8,8a1,15,15a-decahydro-14H-4,6-methanoindolo[3,2,1-ij]oxepino[2,3,4-de]pyrrolo[2,3-h]quinolin-14-ylidene)amino)ethan-1-amine (7).
A solution of Strychnine acid (100 mg, 0.30mmol), ethylenediamine (70 µl, 1.04 mmol) andboric acid (90 mg, 1.45 mmol) in 6 ml of MeOH:CHCl3 (2:4) was stirred for 72 h to room temperature. The reaction mixture was evaporated to dryness under reduced pressure. After, the residue was purified by crystallization from methanol:water (4:1) yielding 55 % of product, m.p. 198-200oC; IR (Vmax, cm-1):
3380 and 3322; 1H NMR (300 MHz, CDCl3) dH: 1.40-1.80 (m, 3H), 2.20-2.30 (m, 2H), 2.68 (m, 1H), 2.72-2.92 (m, 2H), 2.96 (m, 1H), 3.08 (t, 2H, J = 6.44), 3.14-3.62 (m, 5H), 3.64 (t, 2H, J = 6.44), 3.70 (m, 2H), 4.30 (broad, 2H), 4.70-5.84 (m, 2H), 6.70-7.24 (m, 4H) ppm.13C NMR (75.4 Hz, CDCl3) δC: 26.54 (C-17), 28.08(C-7), 29.20(C-18), 39.90(C-14), 40.70(C-27), 45.86 (C-3), 47.94 (C-15), 50.56(C-11), 51.20(C-26), 52.24 (C-13), 59.90(C-16), 64.62 (C-8), 65.00(C-4), 79.18(C-2), 108.50(C-20), 120.00(C-23), 122.00(C-22), 122.86 (C-21), 127.87 (C-9), 134.25 (C-24), 140.3 (C-10), 144.68(C-19),147.76(C-6) ppm. EI-MS m/z: 376.22 (M+ 10). Anal. Calcd. for C23H28N4O: C, 73.37; H, 7.50; N, 14.88; O, 4.25. Found: C, 73.30; H, 7.44.
Synthesis of 2-chloro-N-(2-(((4aR,4a1R,5aS,8aR,E)-2,4a,4a1,5,5a,7,8,8a1,15,15a-decahydro-14H-4,6-methanoindolo[3,2,1-ij]oxwpino[2,3,4-de]pyrrolo[2,3-h]quinolin-14-ylidene)amino)ethyl)acetamide (8)
A solution of 7 (100 mg, 0.26mmol), triethylamine (100 μl, 1.50 mmol) and chloroacetyl chloride (128 μl, 1.60 mmol) in 5 ml of CHCl3was stirring for 48 h to room temperature. The reaction mixture was evaporated to dryness under reduced pressure. After, the residue was purified by crystallization from methanol:water (4:1) yielding 66 % of product, m.p. 84-86oC; IR (Vmax, cm-1): 3318 and 1690; 1H NMR (300 MHz, CDCl3) δH: 1.40-1.86 (m,3H), 2.24-2.30 (m, 2H), 2.70-2.96 (m, 4H), 3.10-3.20 (m, 2H), 3.40 (t, 2H, J = 6.54), 3.54-3.60 (m, 3H), 3.62 (t, 2H, J = 6.54), 3.68 (m, 2H), 4.00 (m, 2H), 4.70-5.83 (m, 2H), 6.70-6.80 (m, 2H), 7.00 (broad, 1H), 7.20-7.28 (m, 2H) ppm. 13C NMR (75.4 Hz, CDCl3) δC: 26.60(C-17), 28.08(C-7), 29.20(C-18), 35.18(C-27), 39.90(C-14), 42.34 (C-31), 45.80(C-3), 47.88(C-15), 50.60 (C-11), 51.30(C-26), 52.18(C-13), 59.90(C-16), 64.56(C-8), 65.00(C-4), 79.27 (C-2), 108.55 (C-20), 120.06 (C-23), 122.00(C-22), 122.86 (C-21), 127.87 (C-9), 134.25 (C-24), 140.30 (C-10), 144.69(C-19), 147.78(C-6), 162.56(C-29) ppm.EI-MS m/z: 452.19 (M+ 11). Anal. Calcd. for C25H29ClN4O2: C, 66.29; H, 6.45; Cl, 7.83; N, 12.37; O, 7.06. Found: C, 66.20; H, 6.40.
Synthesis of 1-((2S)-3((2-(((4aR,4a1R,5aS,E)-2,4a,4a1,5,5a,7,8,8a1,15,15a-decahydro-14H-4,6-methanoindolo[3,2,1-ij]oxepino[2,3,4-de]pyrrolo[2,3-h]quinolin-14-ylidene)amino)ethyl)amino)oxiran-2-yl)naphtalen-2-ol (9).
A solution of 8 (200 mg, 0.44mmol), 2-hydroxy-1-naphthaldehyde (76mg, 0.44mmol), sodium hydroxide (15 mg, 0.37 mmol) in 5 ml of ethanol was stirring for 48 h to room temperature. The reaction mixture was evaporated to dryness under reduced pressure. After, the residue was purified by crystallization from methanol:water (4:1) yielding 85 % of product,m.p. 98-100 oC; IR (Vmax, cm-1): 3402, 3364 and 3324; 1H NMR (300 MHz, CDCl3) dH: 1.40-1.86 (m, 3H), 2.26-2.32 (m, 2H), 2.70-2.96 (m, 4H), 2.98-3.04 (m, 2H), 3.10-3.60 (m, 5H), 3.66 (t, 2H, J = 13.40), 3.69 (m, 2H), 4.06 (broad, 2H), 4.22-4.34 (m, 2H), 4.70-6.86 (m, 4H), 7.12 (m, 1H), 7.20 (m, 1H), 7.26 (m, 1H), 7.28 (m, 1H), 7.30-7.74 (m, 4H) ppm. 13C NMR (75.4 Hz, CDCl3) δC:26.60(C-17), 28.10(C-7), 29.18(C-18), 39.86(C-14), 45.80(C-3), 47.20(C-27), 47.88(C-15), 49.56(C-26), 50.64(C-11), 52.24 (C-13), 59.39 (C-31), 59.90(C-16), 64.62 (C-8), 65.00(C-4), 65.48(C-29), 79.27 (C-2), 108.49(C-20), 116.94 (C-32), 120.00 (C-23), 120.90(C-34), 122.00(C-22), 122.78(C-21), 122.86(C-38), 123.00(C-40), 123.21 (C-37), 126.50(C-39), 127.80(C-9), 128.88(C-41), 130.08(C-35), 132.66 (C-36), 134.20(C-24), 140.28(C-10), 144.68(C-19), 147.76(C-6), 152.40(C-33) ppm. EI-MS m/z: 560.27 (M+ 9). Anal. Calcd. for C35H36N4O3: C, 74.98; H, 6.47; N, 9.99; O, 8.56. Found: C, 74.90; H, 6.40.
Results and Discussion
It is important to mention, that there are many procedures for synthesis of alkaloids derivatives. Nevertheless, despite its wide scope, the former protocols suffer from several drawbacks because some reagents have a limited stability11, 12. Therefore, in this study several straightforward routes are reported for the synthesis of two alkaloids such as brucine and strychnine derivatives. The first step involves preparation of 3 by the reaction of 1 with chloroacetyl chloride using triethylamine as catalyst. The 1H NMR spectrum of 3 shows signals at 1.40-3.22, 3.55-3.62, 3.70, 4.68-5.86 and 7.50 ppm for protons involved in the brucine fragment; at 3.40 and 3.64 ppm for methylene groups present in spacer arm between both imino and chloroacetamide groups; at 3.80, 3.90 ppm for methoxy groups; at 4.00 ppm for methylene of chloroacetamide group; at 7.00 for amide group. 13C NMR spectrum contains peaks at chemical shifts of 26.60-29.20, 39.90, 45.60- 50.58, 52.16, 59.90-147.78 ppm for brucine fragment. Other signals were found at 35.04-51.32 ppm for methylene groups involved in spacer arm between both imino and chloroamide groups; at 42.38 ppm for methylene of chloroacetamide group; at 56.00 and 56.54 ppm for methoxy groups; at 162.56 ppm for amide group.Finally, the presence of compound 3 was further confirmed from mass spectrum which showed a molecular ion at m/z 512.21.
In the second stage is achieved by the synthesis of 4 which have in their chemical structure an oxirane ring. It is noteworthy that many procedures for the formation of oxirane rings are known in the literature, the most widely practiced methods employs some reagents such as Co(III) and Cr(III); However, despite its wide scope, the former protocolshave from several drawbacks such as limited stability of these compounds13,14. In this study, the formation of oxirane ring involved in the compound 4 was made by the reaction of 3 with 2-hydroxy-1-naphthaldehyde in basic medium.The results indicate that 1HNMR spectrum of 4 showed several signals at 1.40-2.94, 3.10-3.60, 3.66, 4.70-5.86 and 7.52 ppm for brucine fragment; at 2.98-3.00 and 3.64 ppm for methylene groups involved in spacer arm between both imino and amino groups; at 3.80 and 3.90 ppm for both methoxy groups; at 4.06 ppm for both amino and hydroxyl groups; at 4.24-4.34 for protons of oxirane ring; at 7.12-7.40 and 7.66-7.70 ppm for both phenyl groups bound to oxirane ring.The 13C NMR spectrum contains peaks at chemical shifts at 26.60-45.82, 50.67-52.24, 59.86-65.00, 79.27-105.70. 127.86, 129.00 and 139.12-147.76 ppm for brucine fragment; at 47.29-49.50 ppm for methylene groups involved in spacer arm between both imino and amino groups; at 55.89-56.60 ppm for both methoxy groups; at 59.30 and 65.78 ppm for carbons of oxirane ring; at 116.88-126.56, 128.92, 130.10-132.66, 152.40 ppm for both phenyl groups bound to oxirane ring. In addition, the presence of compound 4 was further confirmed from mass spectrum which showed a molecular ion at m/z 620.29.
The third stage was achieved by the reaction of Strychnine with ethylenediamine to form an imino group involved in the compound 7. Many procedures for the synthesis of imino groups are described in the literature15,16; nevertheless, in this study boric acid was used as a catalyst, because it is not an expensive reagent and no special conditions for its use are required17. The results indicate that 1HNMR spectrum of 7 showed several signals at 140-2.96, 3.14-3.62, 3.70, 4.70-7.24 ppm for strychnine fragment; at 3.08 and 3.64 ppm for methylene groups of arm bound to both imino and amino groups; at 4.30 ppm for amino group.The 13C NMR spectrum contains peaks at 26.54-39.90, 45.86-50.56 and 52.54-144.68 ppm for carbons of strychnine fragment; at 40.70 and 51.20 ppm for methylene groups; at 147.76 ppm for imino group. Finally, the presence of compound 7 was further confirmed from mass spectrum which showed a molecular ion at m/z 376.22.
The fourth stage involved the reaction of 7 with chloroacetyl chloride to form the compound 8 using triethylamine as catalyst. The 1H NMR spectrum of 8 shows signals at 1.40-3.20, 3.54-3.60, 3.68, 4.70-6.80 and 7.20-7.27 ppm for protons of strychnine fragment; at 3.40 and 3.62 ppm for both amino and imino groups; at 4.00 ppm for methylene group of chloroacetamide fragment; at 7.00 ppm for amide group. 13C NMR spectrum showed peaks at 24.60-29.20, 39.90, 45.80-50.60 and 52.18-144.69 ppm for carbons of strychnine fragment; at 35.18 and 51.30 ppm for both methylene groups bound to imino and amino groups; at 142.30 ppm for methylene group of chloroacetamide fragment; at 144.78 ppm for imino group; at 162.56 ppm for amide group. Additionally, the presence of compound 8 further confirmed from mass spectrum which showed a molecular ion at m/z 452.19.
References
- Dong, Z.;Venkatachalam, M; Weinberg, J;Saikumar, P; Patel, Y. Am. J. Pathol. 2001,158, 1021-1028.
- Takagi, K;Takayanagi, I.Japanese J. Pharmacol.1966,16, 211-216.
- Lazareno, S;Popham,Birdsall, N.J. Med. Chem. 1999,42, 438-445.
- Birdsall, N;Farries, T;Gharagozloo, P; Kobayashi, S;Lazareno, S; Sugimoto, M. Mol. Pharm.1999,55, 778-786.
- Findlay, S.J. Am. Chem. Soc. 1951,73, 3008-3011.
- Záruba, K;Král, V. TetraedronAsym.2002,13, 2567-2570.
- Graham, S; Wemuth, U; White, J. ActaCryst.2006,C62, o353-o357.
- Záruba, K;Králová, J;Rezanka, P;Poucková, P;Veverková, L;Král, V.Org. Biomol. Chem.2010,8, 3202-3206.
- Figueroa-Valverde, L; Díaz-Cedillo, F; López-Ramos, M; García-Cervera, E; Pool- Gómez, E.Asian J. Chem.2012,24, 2173-2176.
- Figueroa-Valverde,L;Díaz-Cedillo, F; López-Ramos, M; García-Cervera, E; Pool- Gómez, E; Torres-Cutz, R.Asian J. Chem.2012,24, 2321-2323.
- Rueping, M; Antonchick, A;Theissmann, T.Ang. Chemie Int. Ed.2006,45:3683-3686.
- Chrzanowska, M;Rozwadowska, M.Chem. Rev. 2004,104: 3341-3370.
- Schaus, S; Brandes, B; Larrow, J; Tokunaga, M; Hansen, K; Gould, A; Furrow, M; Jacobsen, E. J. Am. Chem. Soc.2002, 124:1307-1315.
- Luly, J; Dellaria, J;Plattner, J; Soderquist, J; Yi, N.J. Org. Chem. 1987, 52,1487-1492.
- Shirayev, A;Moiseev, I;Karpeev, S.Arkivok.2005,iv, 199-207.
- Uppiah, D;Bhowon, M;Jhaumeer, S.E-J. Chem.2009,6, S195-S200.
- Figueroa-Valverde, L; Díaz-Cedillo, F; García-Cervera, E; Pool-Gómez, E; Rosas-Nexticapan, M; Ramos-López, M.Asian J. Chem. 2013,25, 6724-26.

![]()




{kind=link}